As a LAMMPS user, I would like to employ the PCFF via the OpenKIM library for a polymer blend. Is there any relevant example available? It is not clear to me how to deal with the bonded interactions of PCFF using OpenKIM.
Thanks,
Evangelos
As a LAMMPS user, I would like to employ the PCFF via the OpenKIM library for a polymer blend. Is there any relevant example available? It is not clear to me how to deal with the bonded interactions of PCFF using OpenKIM.
Thanks,
Evangelos
Hi Evangelos,
We are working on general support for bonded force fields through OpenKIM’s Simulator Model (SM) mechanism for LAMMPS. This includes extensions to the LAMMPS code base to support string labels to identify atom types as an alternative to integer labels used now. This is in final testing and should be available soon. If you would like to send us the LAMMPS commands and parameters for your PCFF model, we can look into setting up an SM - send to [email protected]). Note that for now, support for bonded SMs is limited to LAMMPS.
Best,
Ellad
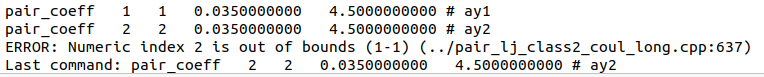
Hello,
Can you please let me know if the following error:
Happening when I only have and need 1 atom type, is due to the string/integer incompatibility between the SM_039297821658_000 potential and LAMMPS.
Thanks in advance,
Best Regards.
Serge
Hi Serge,
IFF is a bonded force field (bond_style, angle_style, etc.) as opposed to a pair_style type potential, so it is handled in a different manner by KIM. This IFF potential has 75 atom types that are defined in its parameter file (https://openkim.org/files/SM_039297821658_000/IFF-supermodel.params). You need to specify this number (75) in your create_box command. You also need to use the appropriate types (as preset in the file linked above). For example, if your simulation contains atoms of IFF type oy1, they would be referred to as atom type 7 in your LAMMPS script. We are in the process of implementing a more flexible approach that would directly use string labels for atom species (like oy1) in LAMMPS input scripts and data files, but that’s not quite ready yet.
Best,
Ellad
Thanks for the clarification Dr. Tadmor.