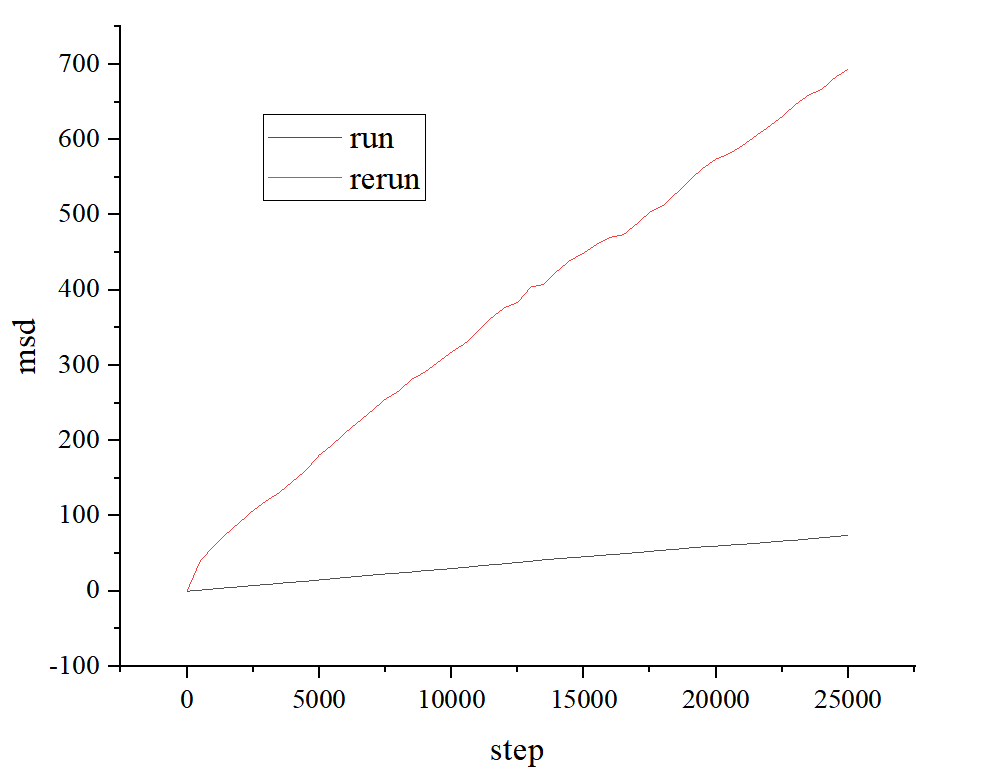
Hello all! I am using the rerun command to calculate the msd(Mean Squared Displacement) and there are some questions. For convience I used the case “LAMMPS 64-bit 8Feb2023\Examples\melt” to describe. I modified the case to output the trace file and msd results
run the “in.melt.lmp”
2.run the “in.meltrerun.lmp”
3.compare the two result
the msd results in 2 methods are differendt and I don’t know how to change my code sice I did’t find the msd calculation with rerun command part
Some discussion have mentioned “read_dump” or “image flag” but I still not clear how to sove this problem.
Your two inputs are not creating output that is consistent with each other due to different settings.
As stated in the compute med documentation it considers atoms passing through periodic boundaries and thus storing only the “in box” coordinates in the dump file is insufficient. You either need to dump unwrapped coordinates or store and read the image flags. Details about that are in the dump command docs
After correcting those two issues, the output is the same except for differences due to the rounding of data in the dump file.
Thank you, now I know how to use "dump 1 all custom 50 dump3.melt id xu yu zu " to dump unwrapped coordinates and rerun the trj file to get the same msd as I wish,but I still be confused by you said “store and read the image flags”, Is this another solution?
A anothe queston is if I only have dump file in “x, y, z”,rather than the unwrapped “xu yu zu”,are there any method to compute the msd with rerun command? I have tested rerun the “dumped x y z” file to dump "xu yu zu ",It gives a wrong unwrapped coordinates compared with "dump xu yu zu " in the run
This should be discussed in the LAMMPS paper. Simply put, coordinates are stored as in-box positions and image flags. The image flag for a direction is incremented or decremented if an atom leaves the box through a periodic boundary to the right or the left, respectively and the atom position reset to the in-box value. The unwrapped positions are reconstructed from in-box positions, image flags, and box length.
No. You cannot use information that is not present.
You can load your trajectory into other software which implement “unwrapping” (the assumption that whenever a particle moves a distance that is more than half the box length, in a single step, it is in fact crossing a box boundary). VMD and MDAnalysis are two examples.