Hello,
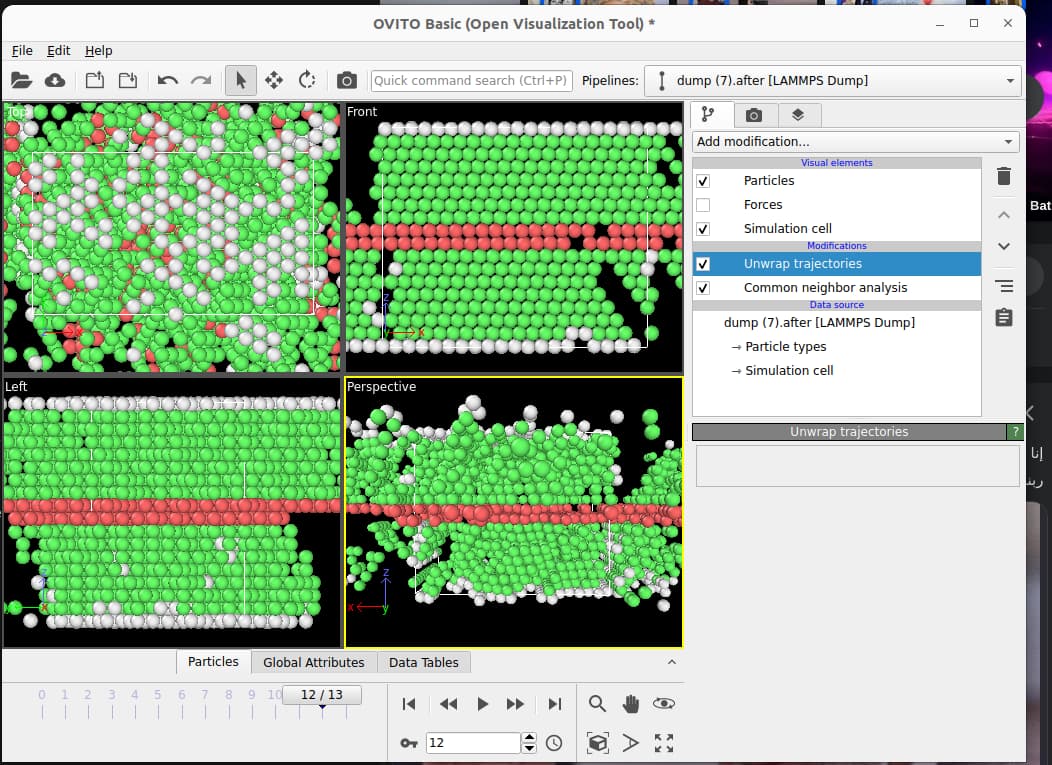
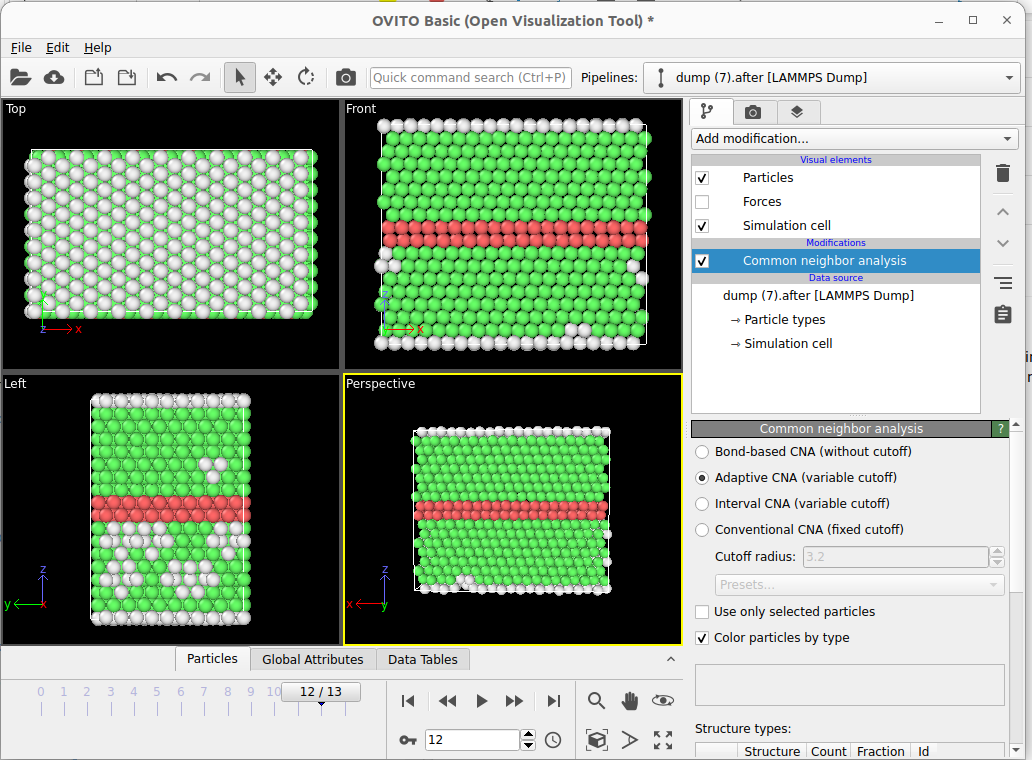
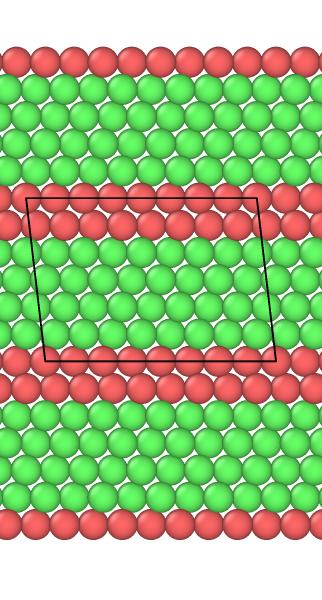
I’m building this code attached to calculate the stacking fault energy and when I visualize using Ovito I find the mismatched grey atoms, how can I avoid them and make it all FCC green atoms except for the stacking fault in red, I’m also attaching the Ovito visualization with and without unwrap trajectories. Also, do I have to make the boundary s along the z direction? Thank you so much for your help!
Best regards,
Dalia
%%writefile NiCrFe.in
# This LAMMPS input script calculates the stacking fault energy of a NiCrFe 60:20:20 alloy.
# The script initializes the system with metal units, 3 dimensions, and periodic boundaries
# in the x and y directions, while being shrink-wrapped in the z direction. It sets the
# lattice parameter and calculates the dimensions of the simulation box.
# Regions are defined for the atoms, and the lattice orientation is specified. Atoms are
# created and distributed in two regions with a 60:20:20 ratio of Ni, Cr, and Fe. The
# initial energy of the system is computed and minimized, and this energy value is stored.
# Atoms in the bottom region are then displaced, and the energy of the system is minimized
# again. The final energy is stored, and the difference between the final and initial
# energies is used to calculate the stacking fault energy, which is then converted to
# mJ/m^2. Finally, the script prints the initial energy, final energy, and stacking fault
# energy and then indicating that the simulation is done.
# ------------------------ INITIALIZATION ----------------------
units metal
dimension 3
boundary p p s
atom_style atomic
variable latparam1 equal 3.52
variable x_displace equal -1*(${latparam1}/sqrt(6))
variable xdim equal ${latparam1}*sqrt(6)/2*10
variable ydim equal ${latparam1}*sqrt(2)/2*10
variable zdim equal ${latparam1}*sqrt(3)*6
# ----------------------- ATOM DEFINITION ----------------------
lattice fcc ${latparam1}
region 1 block 0 ${xdim} 0 ${ydim} 0 16.5 units box
region 2 block 0 ${xdim} 0 ${ydim} 16.5 36.5808 units box
region whole block 0 ${xdim} 0 ${ydim} 0 36.5808 units box
create_box 3 whole # Specify 3 atom types
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
# Create atoms for NiCrFe 60:20:20 alloy in region 1
create_atoms 1 region 1
set type 1 type/fraction 2 0.20 12345
set type 1 type/fraction 3 0.20 12346 # Adjusted to maintain the 60:20:20 ratio
# Create atoms for NiCrFe 60:20:20 alloy in region 2
create_atoms 1 region 2
set type 1 type/fraction 2 0.20 12347
set type 1 type/fraction 3 0.25 12348 # Adjusted to maintain the 60:20:20 ratio
# ----------------------- FORCE FIELDS -----------------------
pair_style eam/alloy
pair_coeff * * /home/dalia/FeCrNi_d.eam.alloy Ni Cr Fe
# ------------------------- SETTINGS --------------------------
group top region 1
group bot region 2
# Compute the initial energy
compute peratom all pe/atom
compute eatoms all reduce sum c_peratom
thermo 1
thermo_style custom step pe c_eatoms
# Minimize energy before displacement
fix 1 all setforce 0 0 NULL
min_style cg
minimize 1e-10 1e-10 1000 1000
# Dump the configuration after minimization before displacement
dump dump_before all custom 1 dump.before id type xs ys zs c_peratom fx fy fz
run 1
undump dump_before
# Store the initial energy
variable initial_energy equal c_eatoms
run 0
variable stored_initial_energy equal ${initial_energy}
# ------------------------- Displacement -----------------------
displace_atoms bot move -1.0 0.0 0.0 units box
# Minimize energy after displacement
fix 1 all setforce 0 0 NULL
min_style cg
minimize 1e-10 1e-10 1000 1000
# Dump the configuration after minimization after displacement
dump dump_after all custom 1 dump.after id type xs ys zs c_peratom fx fy fz
run 1
undump dump_after
# Store the final energy
variable final_energy equal c_eatoms
run 0
variable stored_final_energy equal ${final_energy}
# Calculate the stacking fault energy in eV/Å^2 and convert to mJ/m^2
variable area equal ${xdim}*${ydim}
variable diff_energy equal "v_stored_final_energy - v_stored_initial_energy"
variable stacking_fault_energy equal "v_diff_energy / v_area * 16021.7733" # Conversion factor from eV/Å^2 to mJ/m^2
# Print the energies and stacking fault energy
print "Initial energy: ${stored_initial_energy} eV"
print "Final energy: ${stored_final_energy} eV"
print "Stacking fault energy: ${stacking_fault_energy} mJ/m^2"
# SIMULATION DONE
print "All done"