Dear Lammps user
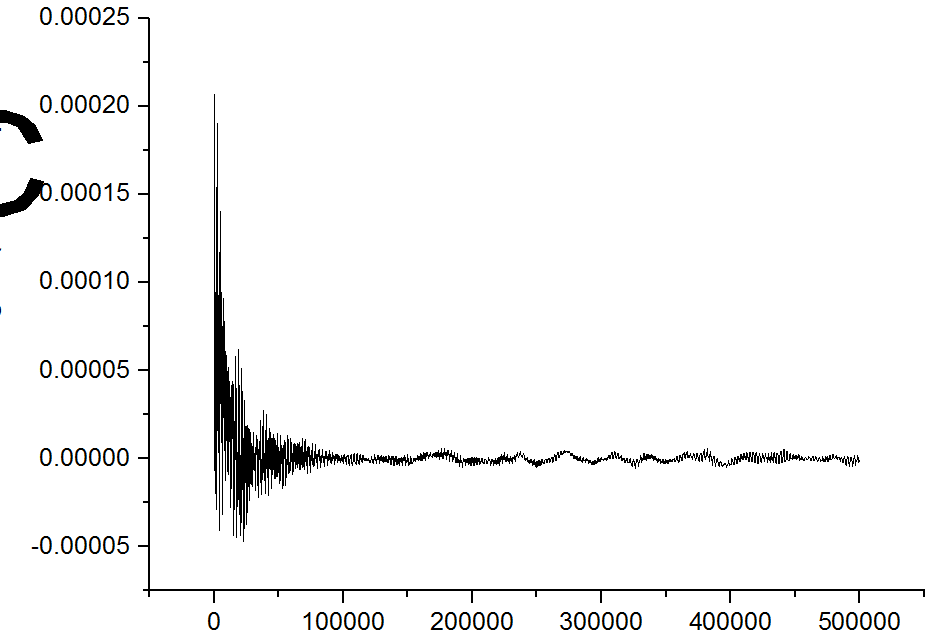
when I use compute vacf to calculate the methane, the output is like white noise as shown in the picture.
“compute vacf all vacf 1 1 1
fix 3 all ave/time 10 10 100 c_vacf[4] file vacf.dat”
Here is the main script:
units real
variable T equal 95
variable Pressure equal 100
variable V equal vol
variable dt equal 0.01
variable p equal 400 # correlation length
variable s equal 1 # sample interval
variable d equal $p*$s # dump interval
variable steps equal 500000
convert from LAMMPS real units to SI
variable kB equal 1.3806504e-23 # [J/K] Boltzmann
variable kCal2J equal 4186.0/6.02214e23
variable A2m equal 1.0e-10
variable fs2s equal 1.0e-15
variable atm2Pa equal 101325.0
variable convertK equal {kCal2J}*{kCal2J}/{fs2s}/{A2m}
variable convertV equal {atm2Pa}*{atm2Pa}{fs2s}*{A2m}{A2m}*{A2m}
boundary p p p
atom_style full
read_data methane0.27.data
pair_style lj/cut/coul/cut 15
bond_style harmonic
angle_style harmonic
pair_modify mix geometric tail yes
special_bonds lj/coul 0.0 0.0 0.5
pair_coeff 1 1 0.066 3.5 # CT
pair_coeff 2 2 0.03 2.5 # HC
pair_coeff 1 2 0.044497191 3 # HC
the remaining parameters are inferred from mixing.
bond_coeff 1 340.0 1.09 # CT-HC
angle_coeff 1 33.0 107.8 # HC-CT-HC
#bond_coeff 1 2845.12 1.09 # CT-HC
#angle_coeff 1 276.14 107.8 # HC-CT-HC
neighbor 1.5 bin
neigh_modify every 10 delay 20 check yes
fix 1 all npt temp $T T 100 drag 0.2 iso {Pressure} ${Pressure} 1000
thermo_style custom step temp pe ke press vol density
velocity all create T 432567 dist uniform
timestep {dt}
thermo $d
run 100000
reset_timestep 0
compute vacf all vacf 1 1 1
fix 3 all ave/time 10 10 100 c_vacf[4] file vacf.dat
run ${steps}
Any useful advice is appreciate.
best ragars!
cheng