I am attempting to study metal hexacyanoferrates using VASP and have been having some issues reproducing DOS calcs from literature but I am unsure where I am going wrong. I am performing the calcs using spin polarization and GGA+U. I have tried both PBE and PBEsol pseudopotentials. All my calculations seem to converge just fine.
My current workflow is to perform a geometric optimization, followed by a single point calc, and then calculate the DOS. I can not upload the INCAR and OUTCAR files but am adding them to this google drive folder. GOOGLE DRIVE FOLDER
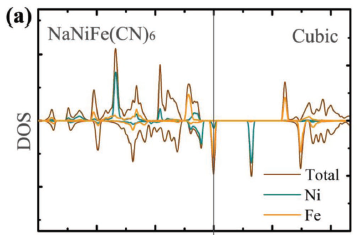
My main concern is that I am missing the energy state around 2 eV. For my calculations I have K+ in the interstitial spacing, but others have shown an energy state near there with potassium as well.
My current workflow is as follows with some input parameters:
Some of the papers that have similar DOS for K2Ni[Fe(CN)6] (or Na instead of K) are below. But also it looks like the system on MAterials Project also has the energy state around 2 eV (https://materialsproject.org/materials/mp-14083/).
Which VASP potentials are you using? We use an old set of pseudopotentials circa ~ 2002 - 2004. We use the K_sv pseudopotential for Potassium as well, which might make a diff.
It could be a difference in U value too, it looks like you’re using 3 eV and 6 eV whereas we use 5.3 eV and 6.2 eV for Fe and Ni respectively. The large difference in Fe U value will definitely have an effect.
Just a quick note that in your INCAR you have:
# Gaussian Smearing
ISMEAR = -5
SIGMA = 0.02
This is not Gaussian smearing, it’s tetrahedral integration (for which the SIGMA value has no effect). Just an FYI.
I am using a running vasp 5.4.4 and the pseudopotentials are from 2007.
With respect to Mkhorton’s comment, I determined that it was in fact the U values. I was using values for U form literature where people presented DOS plots for the same system I am starting with. When I went with U_Fe=3.9 and U_Ni=5.3 there was a large drop in TOTEN and the final DOS had the proper energy band around 2eV. I then ran the system again, changing U_Fe (4.3) and U_Ni (5.7) but keeping the different constant and got the proper system again.
I had one question regarding U and the ability apply values to similar systems. For example, if I optimize the K2Ni[Fe(CN)6] energy with respect to U_Fe and U_Ni, and then substitute in a different metal for Ni but keep U_Fe the same.
My study is on the MnxNi1-x[Fe(CN)6] system. Ni[Fe(CN)6] and the mixed metals are all cubic while Mn[Fe(CN)6] is monoclinic. My idea was to optimize the Ni system, take the U_Fe and fix it while optimize the U for Mn in the monoclinic system, and then use U_Fe, U_Ni, and U_Mn for the mixed metals.
I had one question regarding U and the ability apply values to similar systems. For example, if I optimize the K2Ni[Fe(CN)6] energy with respect to U_Fe and U_Ni, and then substitute in a different metal for Ni but keep U_Fe the same.
There are a lot of differing opinions on this. In principle, U values are element, system and site dependent. Pragmatically, people tend to use fixed U values for specific elements, perhaps varying them depending on what anions are present.
My own opinion on this is that optimizing a U value to fit an experimentally-known band gap is not appropriate, since it is not the “job” of the U to fix the band gap (though, in practice, a good U value will give a DFT-predicted band gap that is closer to its correct value). But finding a good way to fit a U value is difficult.
There are also ways to calculate a U value on a per-site, per-system basis via linear response methods, and this is possible with VASP too (there are tutorials online).
That all makes sense. I will take a stab at optimizing U per the vasp wiki. It seems not too difficult of a procedure. Luckely the systems are rather symmetric.
I guess the last question would be then is there ever coupling or correlation of U values making it not reasonable to independently determine each one.
There’s no easy answer there either. To get a truly self-consistent answer is difficult. I’m personally by no means an authority on this, there’s been some good work on reach self-consistency, e.g. see some discussion here from Timrov et al.
I think, depending on the computational resources you have available, if you’re interested in the DOS I would skip the U altogether and calculate with a higher-level theory like HSE06. There’s still the question of how you get the input geometry however.