Hello.
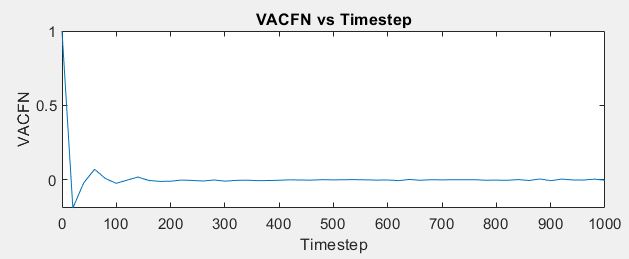
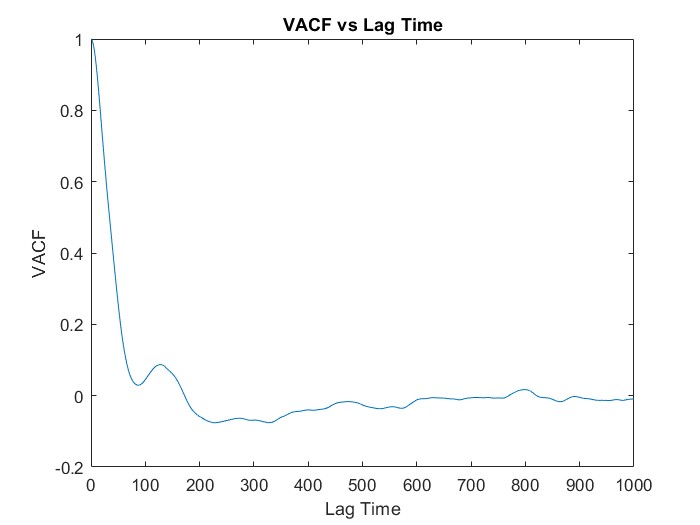
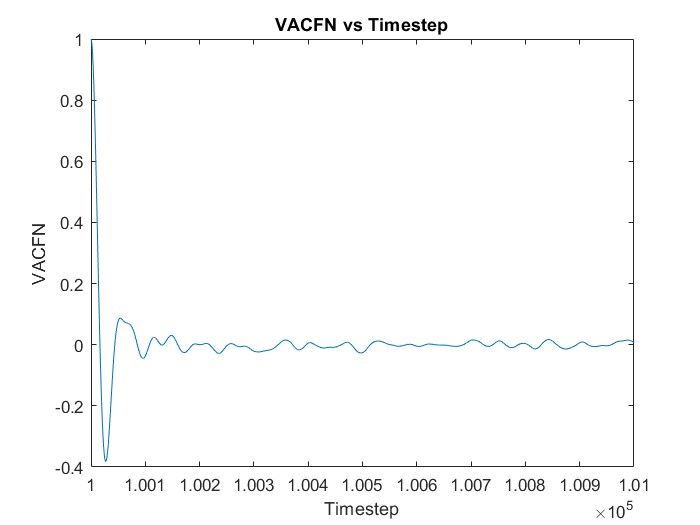
I wanted to calculate VACF for the SPC/E water model where in the input scripts of LAMMPS (attached below), I read the data of the coordinates of all the atoms (9576 atoms total for 3192 molecules) from the GW.dat file and got the values of VACF using “compute” command. However, I got a similar trend of VACF but did not get the accurate curve. I have just started to learn MD and am still a novice, so can anyone tell me how to improve my result even more by looking at the script or give any advise regarding that? Thank you.
###################################
# Simulation settings
###################################
units real
atom_style full
boundary p p p
####################################
# Reading data
####################################
read_data GW.dat
group water type 1 2
group oxygen type 2
group hydrogen type 1
#####################################
# Force interactions
#####################################
pair_style lj/cut/coul/long 10
bond_style harmonic
angle_style harmonic
kspace_style pppm 1.0e-6
pair_coeff 1 1 0.0 0.4000 #H-H
pair_coeff 2 2 0.15535 3.1660 #O-O
pair_coeff 1 2 0.0 1.7753 #H-O
bond_coeff 1 1000 1 # H-O
angle_coeff 1 1000 109.47 # H-O-H
#################################################
# Variables
#################################################
variable Nrun equal 100000
variable Nf equal ${Nrun}/100
variable Ne equal 10
variable Nr equal ${Nf}/${Ne}
variable Ndump equal ${Nrun}/2
variable Nr_rdf equal 0.5*${Nrun}/${Ne}
variable watMoleMass equal 18.0153 # /(g/mol)
variable nAvog equal 6.0221415e23 # Avogadro's number
variable watMoleculeMass equal (${watMoleMass}/${nAvog}) # /(g/molecule)
variable A3_in_cm3 equal 1e-24 # Angstrom^3 in cm^3
variable nAtoms equal atoms
variable nMolecules equal v_nAtoms/3
variable Text equal 298.0
variable Pext equal 1.0
#################################################
# Initial conditions and settings
#################################################
velocity water create ${Text} 1234546 dist gaussian
neighbor 2.0 bin
neigh_modify every 1 delay 0 check yes
##############
# Minimization
##############
minimize 1.0e-5 1.0e-7 1000 10000
##################################################
# Computations
##################################################
reset_timestep 0
timestep 1
#----------- NPT Equilibration -------------------
fix SHAKE water shake 1e-6 200 0 b 1 a 1
fix NPT water npt temp ${Text} ${Text} 100 iso ${Pext} ${Pext} 1500
#fix removeMomentum water momentum 1 linear 1 1 1
compute T water temp
fix TempAve water ave/time ${Ne} ${Nr} ${Nf} c_T
variable P equal press
fix PressAve water ave/time ${Ne} ${Nr} ${Nf} v_P
compute PE all pe pair kspace
variable PE_Mol equal c_PE/v_nMolecules
fix PEAve_Mol all ave/time ${Ne} ${Nr} ${Nf} v_PE_Mol
variable Dens equal v_nMolecules*${watMoleculeMass}/(vol*${A3_in_cm3})
fix DensAve water ave/time ${Ne} ${Nr} ${Nf} v_Dens file wat.dens
thermo_style custom step temp f_TempAve press f_PressAve f_PEAve_Mol f_DensAve
thermo_modify flush yes
thermo 1000
#------------- NVE equilibration ------------------
unfix NPT
#unfix removeMomentum
fix NVE water nve
compute vac all vacf
fix myvcf all ave/time 2 1 20 c_vac[1] c_vac[2] c_vac[3] c_vac[4] file vcf2.dat
run 1000