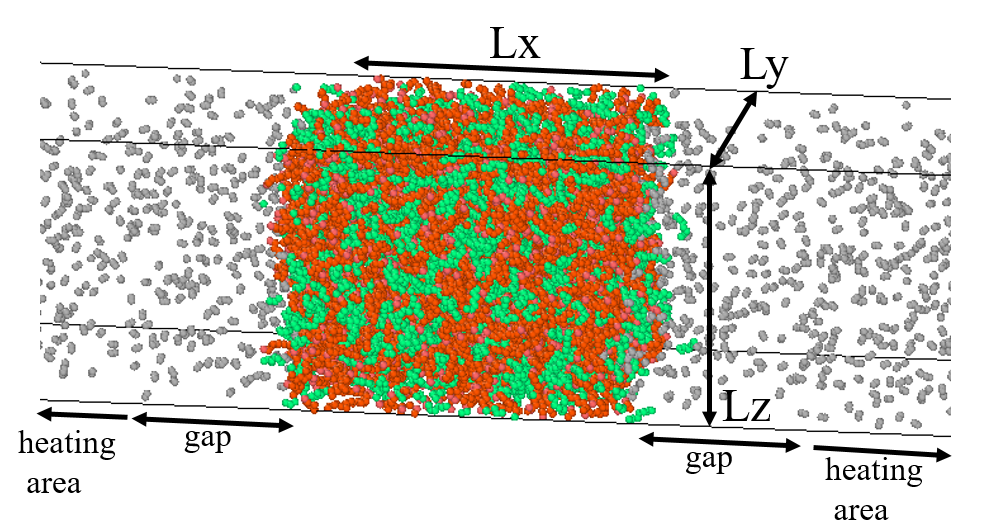
Hi All,
My simulation domain is a cuboid (image attached) with alkane (liquid) at the center and nitrogen as the ambient on either side. One of the cases, for example, involves n-heptane (at 350 K) surrounded by nitrogen at 27 bar and 350 K. When I am creating the particles, I am assigning them a velocity using:
velocity all create 350 492859 rot yes dist gaussian
velocity group_hep zero linear
velocity group_n2 zero linear
I am using the dump command to get the particle locations and velocities:
dump 1 all custom 10000 dump.atom id type x y z vx vy vz mol
Then I have a post-processing code in which I find the mean-spatial velocity variation of the particles. I was expecting that away from the interface and deep into the ambient, the nitrogen gas would follow kinetic theory formulations. But, my code gives a mean velocity of around 725 m/s and a RMS-vel of ~790 m/s. The v-RMS from kinetic theory (sqrt(3KbT/m)) on the other hand is ~570 m/s, on the other hand.
So my questions are:
- Am I thinking on the wrong track? Is the velocity profile from MD not supposed to obey kinetic theory?
P.S.: I have even tried at low pressures of 1 bar and also on single atomic gases like helium, thinking that the shape of the molecule might have been a reason, as kinetic theory is built on assumptions of solid hard spheres as particles. - Is it because of the ensemble that a bias velocity is being imposed on the system at each step and that causes the higher velocity from MD? But I do a “zero linear”, shouldn’t that take care of it?
- My MD is based in united-atom (UA) model. So I model N2 as two N atoms. So with the dump command, I get Vx, Vy and Vz of each N atom. I find the velocity magnitude (sqrt(Vx^2 + Vy^2 + Vz^2) ) for each N atom, sum them up and divide by 2 to get a velocity value for one single N2 molecule. Any flaw in this approach?
Will be hoping to get a response. Thanks in advance.