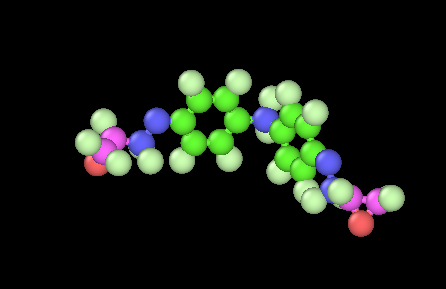
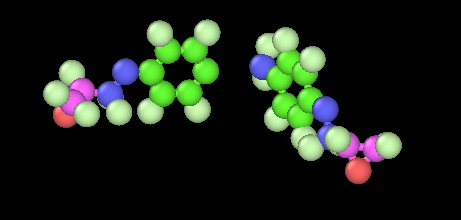I have a high crosslinked epoxy network (90%) that I want to deform until full failure. I am using a “new” reactive forcefield the PCFF-IFF-R that replaces the harmonic bonds with Morse bonds (shifted by a value D) to have the same response near equilibrium distance as the harmonic and slowly dissociate as deformation occures (as the Morse function suggests) . I’m using LAMMPS version June 2023.
When the distance exceeds a certain cutoff (that the forces are relatively small as the derivative of the Morse potential goes to zero) I use the fix bond/break command to break the bonds.
I face some stability issues that I would like some help. I am aware that when a bond breaks, large velocities may ocure locally creating “hot spots” and lost_atoms ERROR or missing _bonds on proc ERROR that as I understand are due to the high forces.
I searches the documentation about the fix temp/rescale and I want to ask if it is suitable to use this command compiled with NPT (that is the ensemble that I perform time integration) and if there is a way to apply the fix tempt/rescale “locally” like to create a group of atoms near a newly broken bond and perform a temp/rescale only to them like the REACTER protocol does with the stabilisation keyword. Any other suggestion on the topic is welcomed! For any further information about the simulation pleas don’t hesitate to ask!
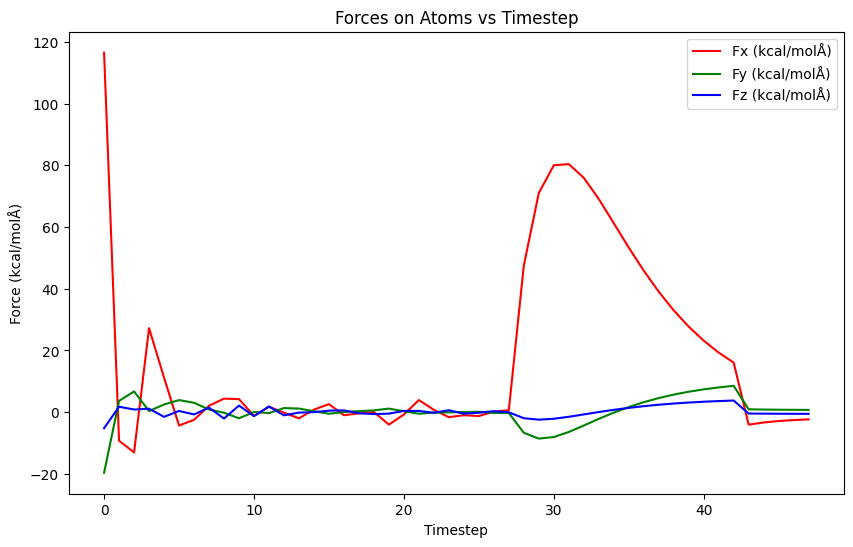
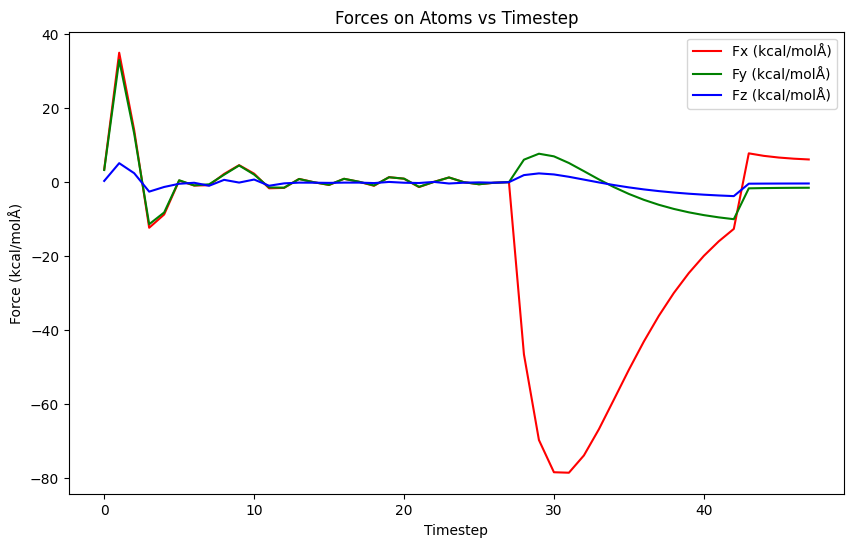
Does this really happen? If what you say is correct, then the bond forces should be very small. The warnings in the documentation are for the more common case in molecular force fields, where you have harmonic bonds that have forces that grow with the distance.
If anything, I would worry about angle and dihedral interactions.
I suggest you construct some typical test geometries with just a monomer and where you move part of it with fix move and immobilize the rest by not using time integration. Then you can output forces and energies (separately for each interactions style) and monitor if there are big changes anywhere.
No. Why would you need that, if can just set up a test system with a small number of atoms and then watch all atoms and have rather short runs until the bond is broken?
Those are two different things.
If you have a smoothly breakable bond potential, that doesn’t do anything for the angle interaction. So that may have still strong forces when the bond breaks. And those will vanish instantly, after you deleted the angle when one of its constituent bonds is deleted. Typically, angle interactions are rather weak, but not all of them. Same for dihedrals.
Whether you are missing angles or bonds mainly is determined by two factors: a) how fast are your atoms moving? and b) how long is your communication cutoff? The communication cutoff is usually sufficient for typical force fields with a non-bonded cutoff in the 12-14 angstrom range.
I can see that when I use fix bond/break the forces are not that big. But the authors of the paper suggest temperature rescaling during the simulation. Is there a way that this can be done under NPT?