Navjot, if this is the fourth duplicate email from me, I apologize. I
had some issues with the size of this email with respect to the
mailing list.
The reason for the density profile being from 0 to 1 is because your
simulation box is triclinic. Since triclinic boxes can be tilted, it's
difficult to divide the box into bins while following the dimensions
of the box. Therefore, the authors of LAMMPS changed it so that the
density profile divides up the triclinic box into bins from 0 to 1.
If you don't need your box to be triclinic, then you could change it
to orthogonal via the change_box command. If your box is not tilted at
all, then the transition from triclinic to orthogonal should be
error-free. Performing a density profile with an orthogonal box does
not have the same distance unit issue as triclinic boxes. With an
orthogonal box, your second column will be in Angstroms (if your units
is metal or real).
This transition should be done before you run the density profile, but
after you load your data. The following command will change the box
from triclinic to orthogonal and remap the atoms such that they fit
within the new box (prevents you from losing any atoms and causing an
error):
change_box all ortho remap
If you want your density profile to be from -100 to +100, then I
recommend changing your simulation box from triclinic to orthogonal
and then running the density profile again.
The commands below should do the job:
change_box all ortho remap
compute solute_x solute chunk/atom bin/1d x center 1 units box
compute solute_y solute chunk/atom bin/1d y center 1 units box
compute solute_z solute chunk/atom bin/1d z center 1 units box
fix x solute ave/chunk 1000 1 1000 solute_x density/mass file
solute1_bin1d_x.txt
fix y solute ave/chunk 1000 1 1000 solute_y density/mass file
solute1_bin1d_y.txt
fix z solute ave/chunk 1000 1 1000 solute_z density/mass file
solute1_bin1d_z.txt
Best Regards,
Will
I tried to cjhange box to ortogonal but i am getting following error
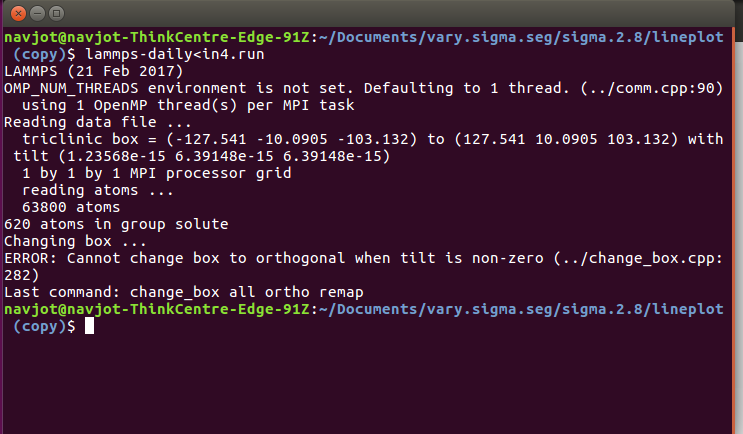
So if your triclinic is tilted at all, you cannot change to orthogonal. You will have to remove the tilt before changing to orthogonal which can be undesirable. From our previous discussions, I think your best option is to leave the simulation box the way it is and increase the number of bins so that the bins go from 0 to 0.005 to 0.01 to 0.015 … to 1.0. The total number of bins would be around 200. Your system setup is from -100 to +100 so if you go with 200 bins your middle point would be exactly 0.50. In your case, it does not matter if the density profile matches your simulation box dimensions. So -100 would correspond to 0.0 and +100 would correspond to 1.0. 0 would be 0.5.
tilt factors of the order of magnitude of 1.e-15 are effectively zero. their size is of the magnitude of double precision floating point precision limits. so the question is, why write out a triclinic style data file at all? an there is no need to use change_box. a smart person would just delete the line with the three tilt factors from the data file and it will magically become orthogonal.
axel.
I increased number of bins to 200 and got following curve,which show segregation at 0.5 which is grain boundary position. So what should I write for x and y axis labels along with units.
I am using LJ units.
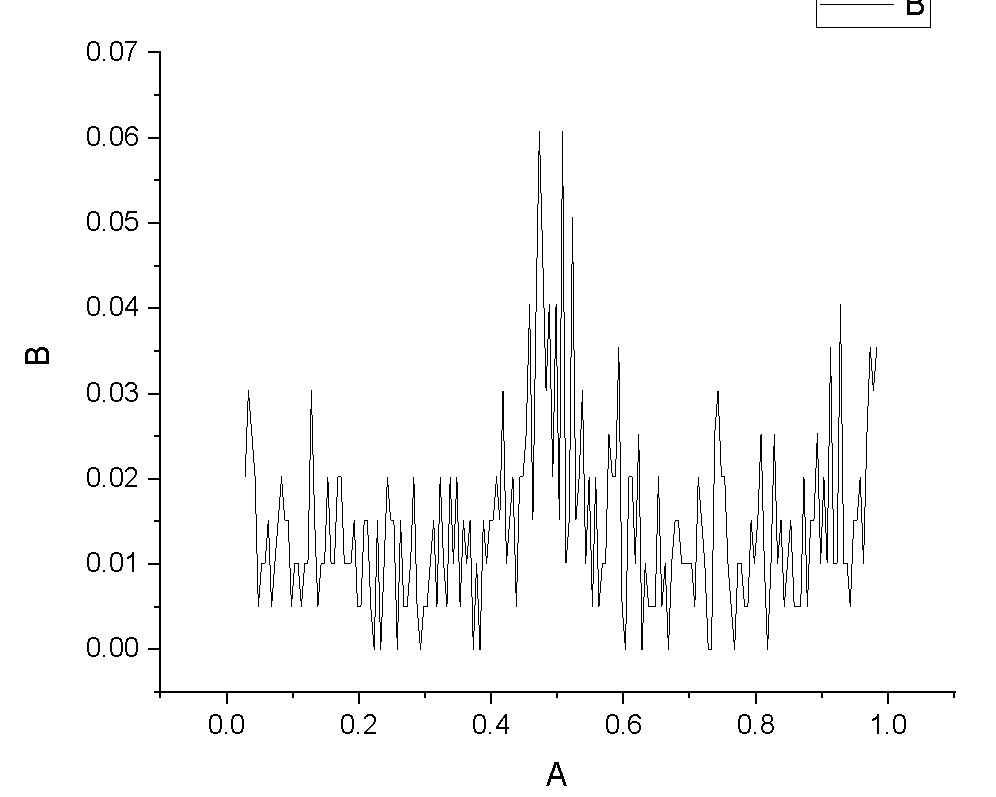
Glad you got it working. For y: concentration of Ni atoms. For x: bin position. Per the documentation, “For style lj, all quantities are unitless.” (https://lammps.sandia.gov/doc/units.html)
Will
Than you so much for the help and explanation.
I have just last question that for y axis concentration should have units of mass/volume. I didn’t understand the statement “For style lj, all quantities are unitless”
You’re welcome! If your units style is lj, all quantities are unitless. I’m not familiar with transforming unitless quantities into quantities with units in LAMMPS because I always use real units. If you don’t have a particular need for using lj units, I recommend using almost anything else (I prefer real units).
I need to use lj units because I am using LJ potential.
Can I use real or metal units even if I am using LJ potential?
Yes. The units are independent of the potential you’re using.
I used real units,so y axis has a units of grams/cm^3.I need to express concentration on y axis in terms of percent. I try to google that and I got some chemistry terms which I don’t know.
Actually my solvent is Al and solutes are Ni. I want to show what % of Ni are segregated in grain boundary as compared to neighboring grains.
If you need percent, you could try density/number which should give you number of Ni atoms in each bin. Since you know the total number of Ni atoms, simply divide number profile by the total number of Ni atoms to get percentage of Ni atom.
I calculate percentage of Ni atoms by diving density/number by total number of Ni atoms (which was 620). I got values in order of 10^-6. I was wondering is it logical to get such small numbers.
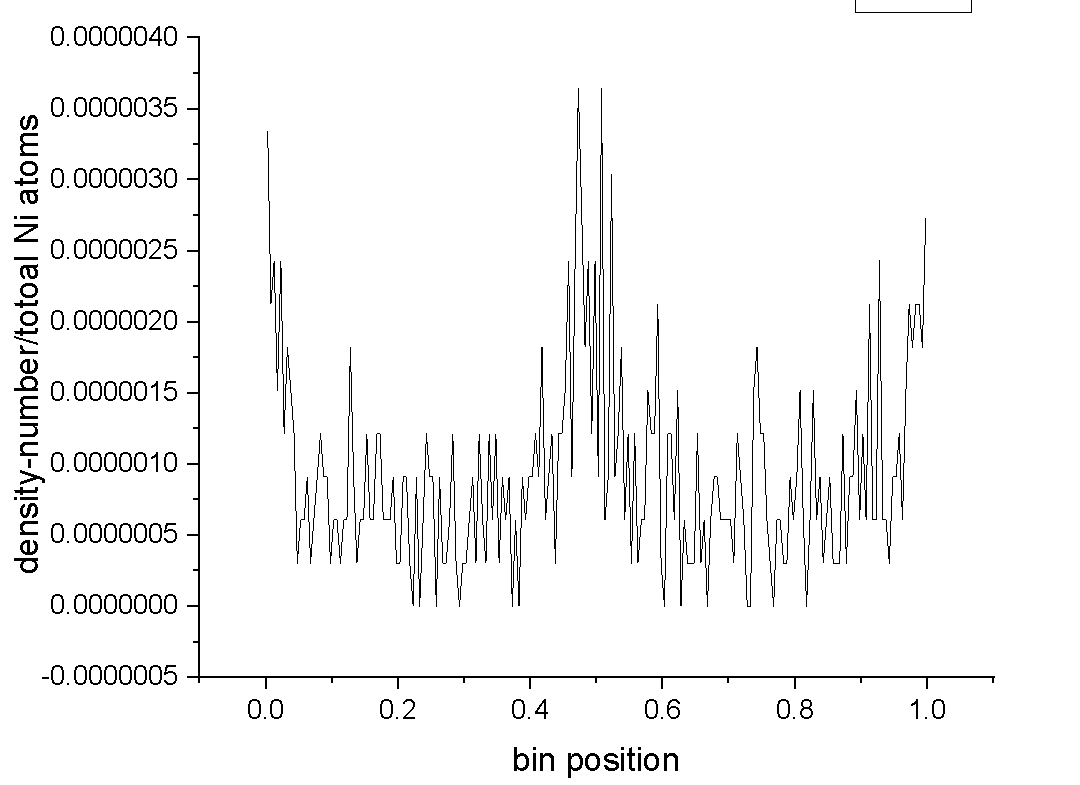
Perhaps density/number already gives you concentration in percent? I’m not sure. Maybe someone with more expertise could chime in?