Dear Lammps users,
I am using compute Voronoi/atom option of Lammps to obtain the occupancy of the atoms relating to the minimized structure. I am running NVT ensemble on the simple cubic Al system with periodic boundary condition. After doing minimization, I delete 1% of atoms randomly to make a system with initial 1% vacancy. The temperature is kept around 700K.
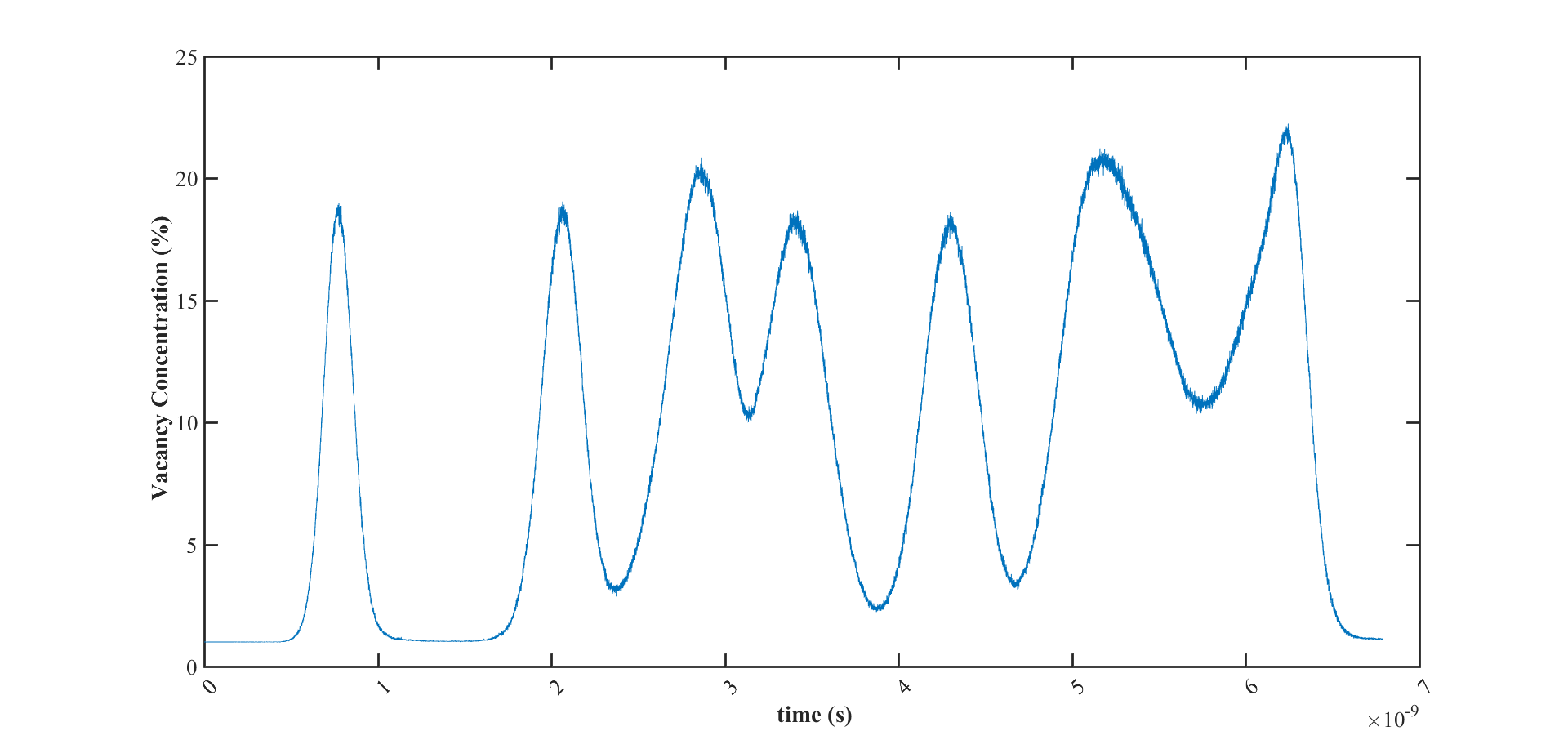
The below figure is the variation of vacancy concentration versus time. It shows that the initial vacancy concentration is equal to 1% which is correct and I applied it manually but after less than 1ns the vacancy concentration reaches around 20%.
It is strange for me that the vacancy concentration can reach up to 20% (while the system has constant volume) at fcc structure that is already a closed pack structure.
Is that possible that some of the atoms have overlap or there is something in voronoi calculation that can cause this huge vacancy concentration?
Thanks,
Sara
Dear Lammps users,
I am using compute Voronoi/atom option of Lammps to obtain the occupancy of the atoms relating to the minimized structure. I am running NVT ensemble on the simple cubic Al system with periodic boundary condition. After doing minimization, I delete 1% of atoms randomly to make a system with initial 1% vacancy. The temperature is kept around 700K.
The below figure is the variation of vacancy concentration versus time. It shows that the initial vacancy concentration is equal to 1% which is correct and I applied it manually but after less than 1ns the vacancy concentration reaches around 20%.
It is strange for me that the vacancy concentration can reach up to 20% (while the system has constant volume) at fcc structure that is already a closed pack structure.
Is that possible that some of the atoms have overlap or there is something in voronoi calculation that can cause this huge vacancy concentration?
what do you see, when you visualize your trajectory? do atoms stay in
place, or are they moving around? can you confirm that you have no
center of mass translation of the total system?
you say you have a temperature of 700K, which should already cause
some motion despite being below the melting point. apropos, what is
the melting point determined for the potential you are using?
axel.
Atoms are moving inside the box, they are changing their position. The simulation box stays at the same place and just atoms are moving. Then I do not have any centre of mass translation.
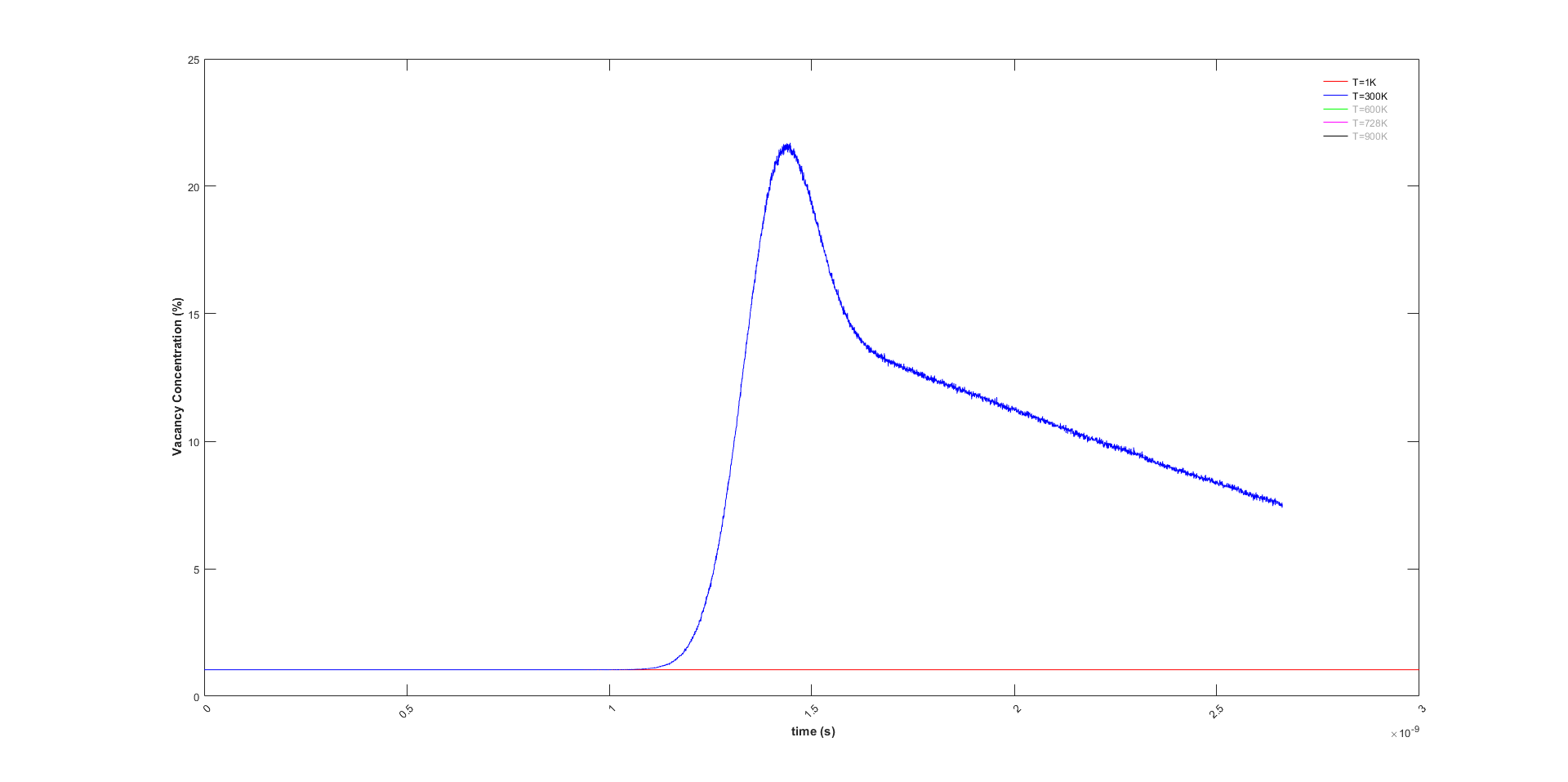
The melting temperature is around 900K. I have tried different temperature. I saw the same behaviour even at 300K. Just at 1K, I do not see any change at vacancy concentration, which makes sense since atoms do not have the thermal vibration(see figure below).
Atoms are moving inside the box, they are changing their position. The simulation box stays at the same place and just atoms are moving. Then I do not have any centre of mass translation.
The melting temperature is around 900K. I have tried different temperature. I saw the same behaviour even at 300K. Just at 1K, I do not see any change at vacancy concentration, which makes sense since atoms do not have the thermal vibration(see figure below).
this is not unexpected. please make certain you are fully aware of
what the occupation keyword does by re-reading the corresponding
documentation carefully.
essentially, you are generating the idealized voronoi tesselation at
the beginning of the simulation and then compare how current atom
positions fit into those.
if there are reconstructions and significant vibrations, some of the
original voronoi cells will be occupied by multiple atoms.
axel.
Yes, I made the minimized structure as the reference configuration for the Voronoi calculation (considering it as an idealized structure). I know that at each time step the occupancy number that I get (as the first column of the Voronoi/compute occupation output) applies to the atomic sites of the reference configuration. Then it computes the number of atoms that occupy each voronoi site and the occupation is the number of atoms sitting on each voronoi site of the reference configuration. Then If I just filter those atoms with Occupancy=0 (sites that are occupied by zero atoms), they can represent vacancy. I just calculate the percentage of these vacancy sites (the summation of atoms with occupancy=0/total number of atoms%100) versus NVT time.
Then having 20% of vacancy means that I have an Al fcc structure with 1% initial vacancy concentration, wich under NVT ensemble (fix Volume and number of atoms) it gets 20% porous? I can believe that it can go to 2% or 3% but 20% is too much for a structure that is already closed pack. Does voronoi calculation consider the atomic radius?
1 Like
Yes, I made the minimized structure as the reference configuration for the
Voronoi calculation (considering it as an idealized structure). I know that
at each time step the occupancy number that I get (as the first column of
the Voronoi/compute occupation output) applies to the atomic sites of the
reference configuration. Then it computes the number of atoms that occupy
each voronoi site and the occupation is the number of atoms sitting on each
voronoi site of the reference configuration. Then If I just filter those
atoms with Occupancy=0 (sites that are occupied by zero atoms), they can
represent vacancy. I just calculate the percentage of these vacancy sites
(the summation of atoms with occupancy=0/total number of atoms%100) versus
NVT time.
Then having 20% of vacancy means that I have an Al fcc structure with 1%
initial vacancy concentration, wich under NVT ensemble (fix Volume and
number of atoms) it gets 20% porous? I can believe that it can go to 2% or
3% but 20% is too much for a structure that is already closed pack. Does
voronoi calculation consider the atomic radius?
no. that is actually one of its appealing features. please look up
elsewhere what voronoi tesselation does. it is required to correctly
interpret the results.
i think you are misinterpreting the data and the process. what you are
doing is the following.
at the very beginning, you divide the total volume of your simulation
cell into small subvolumes, each of which is assigned to exactly one
atom.
during the run, you count how many atoms are in each subvolume. there
are multiple ways that you can have (temporarily) a significant number
of unoccupied voronoi cells:
- large thermal fluctuations (especially if the system is not yet well
equilibrated)
- change of the lattice structure (that would not be oscillatory, though).
- large amplitude phonons (the initial graph you have shown, looks
like it could represent two such phonons passing through your system
at different speeds).
- center of mass translation (flying icecube).
rather than only looking at cells with zero occupancy, you should also
check for cells with 1, 2, or more atoms in them, particularly 2.
axel.
OK. Then, if I do not get it wrong, I should not interpret this parameter as a direct measurement of vacancy concentration (or the porosity of material). It just shows that locally, how many of these subvolumes are unoccupied while some of them can be occupied by 2 or 3 atoms. (like the figure below)
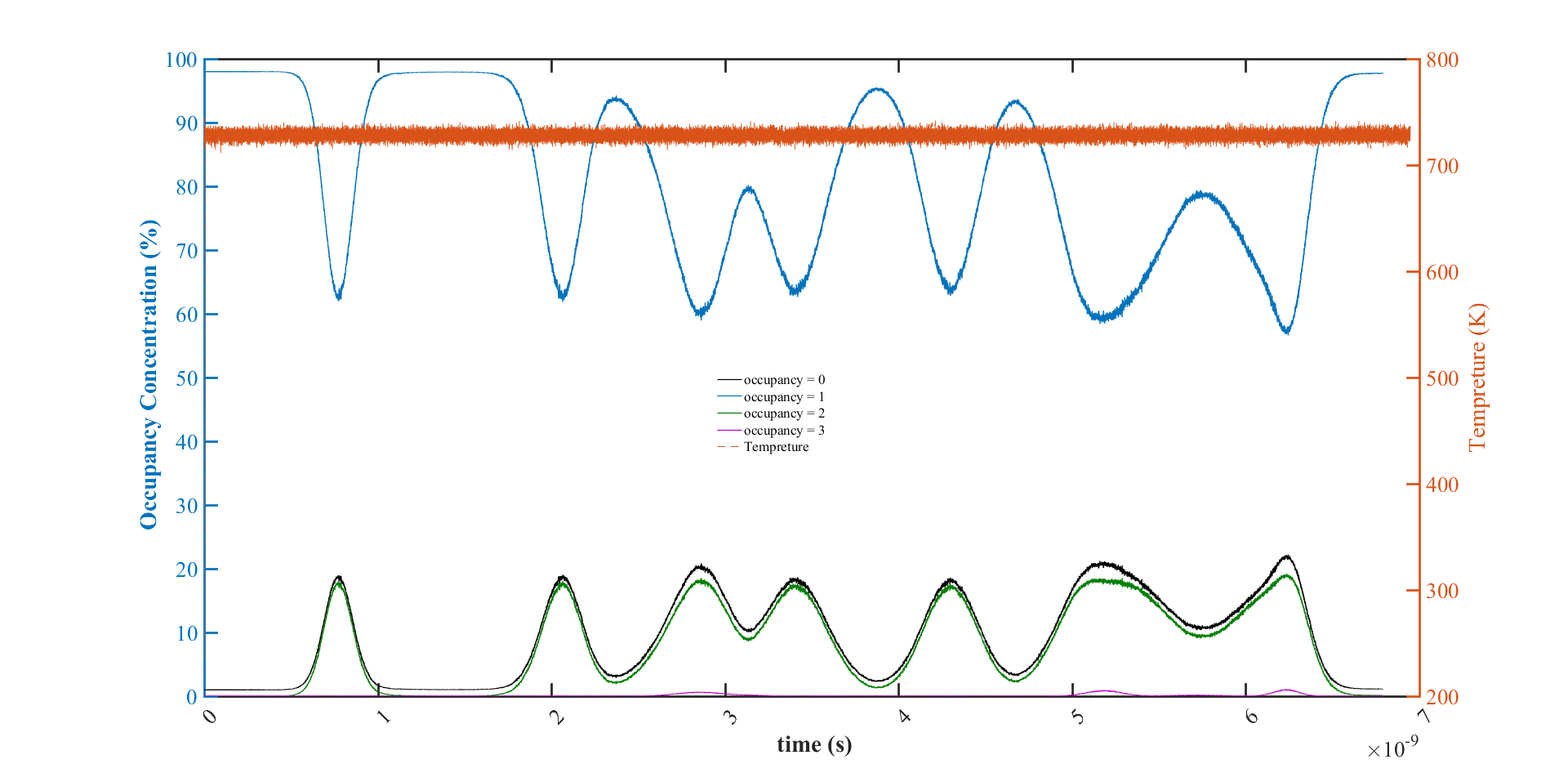
Then about temporarily a huge number of unoccupied voronoi cell in my system, I do not think that thermal vibration is the main reason (the temperature plot does not show that much temperature change, above figure). I think due to the oscillatory behaviour of the graph, it is most probably by the thermal photon. Is there any way that I can control or damp this oscillation?
Thanks,
Sara
OK. Then, if I do not get it wrong, I should not interpret this parameter as a direct measurement of vacancy concentration (or the porosity of material). It just shows that locally, how many of these subvolumes are unoccupied while some of them can be occupied by 2 or 3 atoms. (like the figure below)
Then about temporarily a huge number of unoccupied voronoi cell in my system, I do not think that thermal vibration is the main reason (the temperature plot does not show that much temperature change, above figure).
you are misunderstanding what thermal fluctuations are: they are local
position changes due to high kinetic energy, not changes in
temperature (those would be called temperature fluctuations).
I think due to the oscillatory behaviour of the graph, it is most probably by the thermal photon. Is there any way that I can control or damp this oscillation?
what is happening should be visible from visualizing your trajectory
in sufficient resolution. what would be a suitable countermeasure
depends on your initial geometry, your input settings and
equilibration procedure. impossible to say from just a couple of
analysis graphs. i don't want to give bad advice based on a guess of
the most common problems that people have.
at this point, i would recommend to talk to somebody local with
expertise in these things, usually that would be your adviser or some
experienced group member or some collaborator of your group. this is
more a question of knowing and applying general MD simulation best
practices and proper understanding of them. this is beyond the scope
of this mailing list.
axel.
Assuming you did everything else right, I’ve seen cases where NVT leaves a lot of empty space because I didn’t equilibrate the model properly. Removing even 1 percent of atoms can cause quite a ruckus. Try equilibrating it differently, keeping the same density for 700K as in the perfect crystal (around 0 K ) is quite demanding. You could try NPT and then perhaps there’s a way to measure vacancy for the volume containing the original Voronoi cells with respect to the original model by creating a sub-group of your atoms. Nonetheless, this is a shot in the dark without more information about the physical process you’re trying to model.