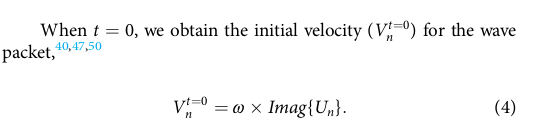Hi all,
I generated a Gaussian wave packet with a specific velocity on a CNT structure. The wave packet was generated on an energy-minimized structure. During the NVE ensemble, the wave packet is not propagating. Instead the amplitude keeps increasing during the NVE integration, though the initial wave packet has a fixed amplitude. May I know the reason behind this issue ?
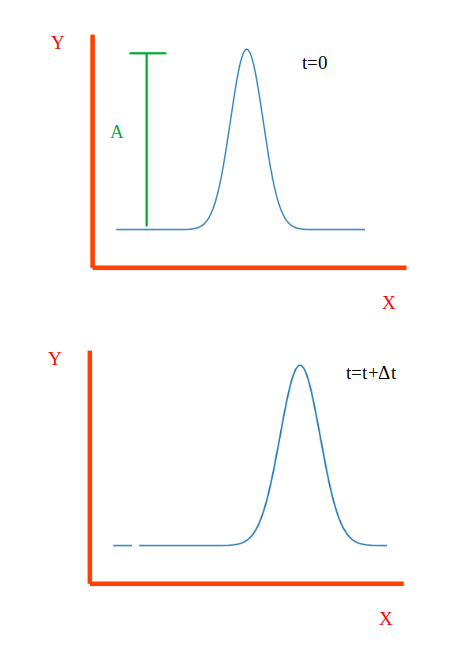
To make my question clearer, I’d like to rephrase it. Consider a scenario where a 1-dimensional chain of atoms is displaced with an amplitude of A=0.01pm at t=0, as shown in the figure.
I want to propagate this wave along the x-direction. The issue I am encountering is that when I apply the velocities Vx=Ux⋅ω and Vy=Uy⋅ω (with ω=10 THz, Ux and Uy are the displacement of atoms along x and y -direction), the wave does not propagate as expected. Instead, it starts spreading from its initial position. For time integration, let’s consider the NVE ensemble. In LAMMPS, how should I specify the velocities to propagate the wave along the x-direction as shown in the figure?
(Note: I initially tried to propagate a wave along a CNT channel. Based on those observations, I simplified the problem for better understanding. I have not yet attempted the 1-D simulation discussed in the rephrased question. The question concerns how to specify the velocities to propagate a wave.)
Your script almost certainly does not do what you think you want to do. (It is impossible to discuss further more definitively unless you show us the script you have written and started using.)
Quite apart from script correctness, an MD simulated material will almost certainly have some non-zero dispersion (that is, different frequencies of the wave will move at different speeds), making coherence unlikely. But that is a later problem, and not a LAMMPS problem at all.
Dear @srtee !
Here I have provided the LAMMPS scripts that use a simple Lennard-Jones (LJ) model to propagate a Gaussian wave packet along the z-direction. As anticipated, the wave does not propagate.
Thanking you,
With regards,
Vivek
# LAMMPS Input Script for a 1D Atomic Chain with gaussian wave packet
units lj
dimension 3
boundary p p p
atom_style atomic
# # ****************** simulation box ********************* #
region box block -5 5 -5 5 0 100 # Create a box with dimensions along z-direction (20 atoms)
create_box 1 box
mass 1 1.0
variable spacing equal 1.0
create_atoms 1 single 0 0 0
variable i loop 100
label loop
variable zpos equal ${i}*${spacing}
create_atoms 1 single 0 0 ${zpos}
next i
jump SELF loop
# ****************** gaussian wave packet input ********************* #
variable amplitude equal 1.0
variable sigma equal 5.0
variable x0 equal 50.0
variable freq equal 5
# ****************** gaussian wave packet generation ***************** #
variable j loop 101
label loop_start
variable x equal ${j}-1
variable gaussian equal ${amplitude}*exp(-(((${x}-${x0})^2)/(2*${sigma}^2)))
variable vel_vy equal ${gaussian}*${freq}
set atom ${j} x 0.0 y ${gaussian} z ${x}
set atom ${j} vx 0.0 vy -${vel_vy} vz 0.0
next j
jump SELF loop_start
# ****************** gaussian wave packet end ***************** #
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0
neighbor 0.3 bin
neigh_modify every 1 delay 0 check yes
timestep 0.01
thermo 100
fix 1 all nve
dump save_cor all custom 1 atom.data id x y z
dump_modify save_cor sort id
run 1000
# ********************************************************************** #
The use of the term “wave packet” is a complete misnomer here.
What you are modeling is not a wave packet where you propagate the wave function and then compute its density by taking the square. Instead you have a string of interacting atoms.
How is this unexpected? The atoms will try to minimize the forces between them. Have you checked what happens without adding a velocity?
If you want the string of atoms move at constant velocity without changing their relative positions you must remove the forces on those atoms. Or just skip the whole process and just use fix move. In both cases the question remains of what the purpose of such a calculation is.
Dear @akohlmey !
Thank you for your response. To provide better context, I am including the reference article I am basing my simulation on.
A phonon wave packet study of thermal energy transport across functionalized hard-soft interfaces. DOI: 10.1063/1.5095775
According to the authors, the wave packet (displacement of atoms Un) can be generated using the following equation:
The initial velocity condition is,
The authors mentioned that once the velocity is specified, the wave packet will propagate during NVE integration. I have used Equations (1) and (4) to create the wave packet and the initial velocity for the system, but it hasn’t resolved the issue. Here, I have modified my LAMMPS script which considers the wave number (k0) also.
# LAMMPS Input Script for a 1D Atomic Chain with gaussian wave packet
units lj
dimension 3
boundary p p p
atom_style atomic
# # ****************** simulation box ********************* #
region box block -5 5 -5 5 0 100 # Create a box with dimensions along z-direction (20 atoms)
create_box 1 box
mass 1 1.0
variable spacing equal 1.0
create_atoms 1 single 0 0 0
variable i loop 100
label loop
variable zpos equal ${i}*${spacing}
create_atoms 1 single 0 0 ${zpos}
next i
jump SELF loop
# ****************** gaussian wave packet input ********************* #
variable PI equal 3.141592653589793
variable amplitude equal 1.0
variable sigma equal 5.0
variable x0 equal 50.0
variable freq equal 5
variable k0 equal 50.0
# ****************** gaussian wave packet generation ***************** #
variable j loop 101
label loop_start
variable x equal ${j}-1
# Real part of wave packet equation
variable gaussian equal ${amplitude}*exp(-(((${x}-${x0})^2)/(2*${sigma}^2)))*cos((2*${PI}/100)*(${x}-${x0})*${k0})
# Imaginary part of wave packet times frequency
variable vel_vy equal ${gaussian}*${freq}*sin((2*${PI}/100)*(${x}-${x0})*${k0})
set atom ${j} x 0.0 y ${gaussian} z ${x}
set atom ${j} vx 0.0 vy ${vel_vy} vz 0.0
next j
jump SELF loop_start
# ****************** gaussian wave packet end ***************** #
pair_style lj/cut 2.5
pair_coeff 1 1 1.0 1.0
neighbor 0.3 bin
neigh_modify every 1 delay 0 check yes
timestep 0.01
thermo_style custom step temp pe ke etotal
thermo 100
fix 1 all nve
dump save_cor all custom 1 atom.data id x y z
dump_modify save_cor sort id
dump save_vel all custom 1 atom_vel.data id vx vy vz
dump_modify save_vel sort id
run 1000
# ********************************************************************** #
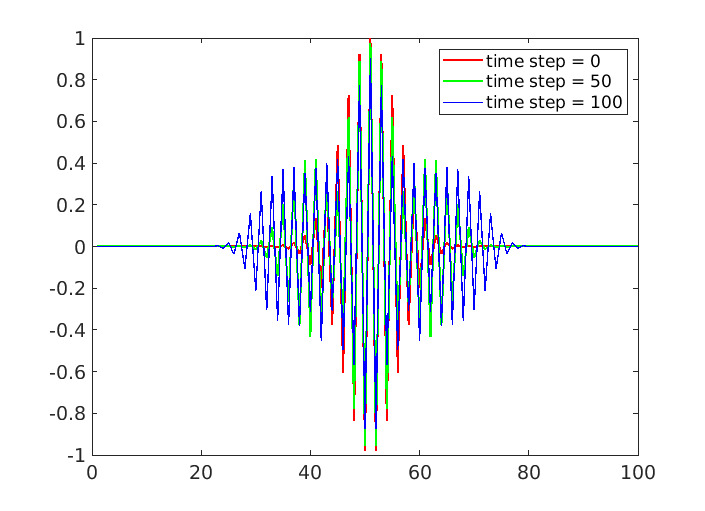
Further I plotted my simulation results at various time steps for your reference.
From the above figure it is visible that the wave is not propagating along z-direction. I wonder what causing this issue. Also without any initial velocity I still face the same.
This is not a LAMMPS issue, but an issue of the science and - while I am not an expert in these matters - your input appears wrong in a way that there seems to be a fundamental lack of understanding on your side. If you want to set up a phonon in a 1-d system you should only have assigned positions and velocities in this one dimension, everything else should be 0.0. Yet, you are changing the y values and positions. In stead the phonon would be represented by a changing in density in z, i.e. atoms would be closer together and further apart.
There is not much that we can do for you from the LAMMPS side, since your problems are of the conceptual kind. LAMMPS will do what you tell it to do, but what you tell it is effectively your problem and something that you need to discuss with those that know and care about your research.
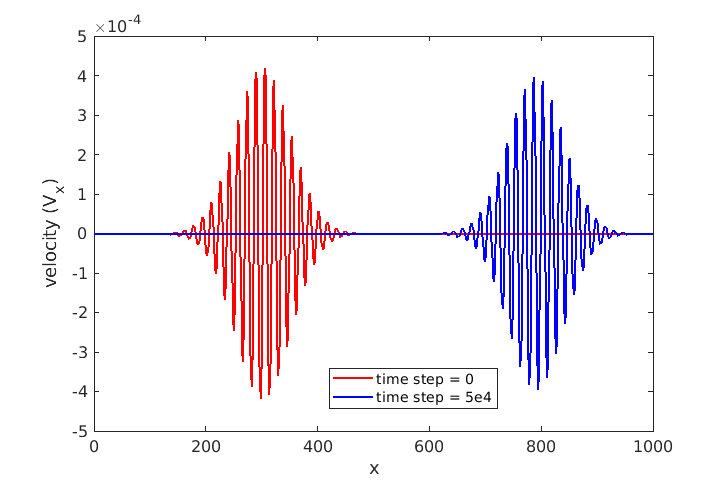
Dear all !
Finally, the issue has been resolved after a discussion with Dr. Xingfei Wei, one of the authors of the aforementioned paper. I identified that the issue was with my initial velocity and the respective wave vectors, which were not assigned according to the phonon dispersion of a 1D atomic chain. With the proper initial conditions, the wave packet is now propagating correctly. Here, I share my updated LAMMPS script and system file. These may be helpful to those whose research interests align with this study.