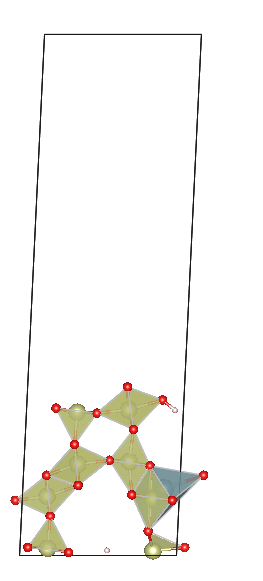
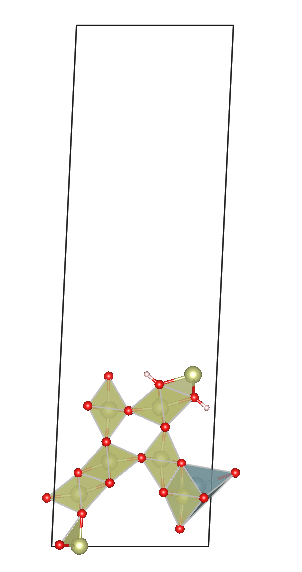
Hello dear pymatgen users! I’ve been using SlabGenerator in pymatgen to obtain slabs. My bulk structure is quite complex, with many atoms not aligned in a single layer (possibly termed corrugated layers). From what I understand, pymatgen generates slabs layer-by-layer (the atoms in layer should be very close together), not atom-by-atom. So, what criteria does pymatgen use to define these atoms as a single layer? Can users modify this criterion?
Here’s my specific code (ignore the pseudo uranium atom addition, it’s just to break symmetry), along with my BULK file. My input parameters are 1 0 0 1 1 1, meaning Miller indices of (100), no supercell operation, and cutting a slab with one interlayer distance thickness. I’ve also attached images of two adjacent slabs where pymatgen have moved more than one atom at a time.You can see that new slab added three atoms. In other words, what should I do if I want to get one slab between these two slabs?
I’d be very grateful for any responses. Thank you!
import sys
import os
from pymatgen.core.surface import SlabGenerator
from pymatgen.core import Structure
"""
1.Read user input parameters
"""
if len(sys.argv) < 7:
print("The parameter is insufficient. Usage method: python script.py m k l a b thickness")
sys.exit(1)
m, k, l = int(sys.argv[1]), int(sys.argv[2]), int(sys.argv[3])
a, b = int(sys.argv[4]), int(sys.argv[5])
thickness = float(sys.argv[6])
#Create and go to the mkl folder
mkl_folder = f"{m}{k}{l}"
if not os.path.exists(mkl_folder):
os.makedirs(mkl_folder)
os.chdir(mkl_folder)
"""
2.Use pymatgen to generate slab structure.
"""
#1. Add pseudoatom U to break the symmetry
initial_structure = Structure.from_file("../CONTCAR") # Attention path!
initial_structure.append("U", [0.7, 0.8, 0.9], coords_are_cartesian=False)
initial_structure.to(fmt="poscar", filename="CONTCAR_with_U.vasp")
#2. Perform the function
# Load the CONTCAR file to create the initial structure
initial_structure = Structure.from_file("CONTCAR_with_U.vasp")
# Define the Miller index
miller_index = (m, k, l)
# Calculate the crystal face spacing
d_spacing = initial_structure.lattice.d_hkl(miller_index)
d_spacing = d_spacing - 0.01
# Create a SlabGenerator instance
slabgen = SlabGenerator(initial_structure, miller_index=(m, k, l),
min_slab_size=d_spacing * thickness,
min_vacuum_size=15.0,
in_unit_planes=False)
# Get slab
slabs = slabgen.get_slabs()
slab_files = []
# Save the slab structure to the file
for i, slab in enumerate(slabs):
# Enlarge the cell(If need)
supercell_slab = slab * (a, b, 1)
filename = f"CONTCAR-out{i+1}.vasp"
supercell_slab.to(fmt="poscar", filename=filename)
slab_files.append(filename)
This is my bulk file:
Potassium iridium dioxide (0.25/1/2)
1.0
10.6568880081 0.0000000000 0.0000000000
0.0000000000 3.1951665878 0.0000000000
0.4598528482 0.0000000000 9.6042059680
Ir O H
8 16 2
Direct
0.358675063 0.000000000 0.176839471
0.643276751 0.000000000 0.829865575
0.856723309 0.500000000 0.670134425
0.141324937 0.500000000 0.323160529
0.842498183 0.000000000 0.342902631
0.155891865 0.000000000 0.650927007
0.344108135 0.500000000 0.849072993
0.657501817 0.500000000 0.157097369
0.164995074 0.000000000 0.188101739
0.833402395 0.000000000 0.803040028
0.666597664 0.500000000 0.696959913
0.335004926 0.500000000 0.311898261
0.779002905 0.000000000 0.148541719
0.212640449 0.000000000 0.862887204
0.287359536 0.500000000 0.637112856
0.720997095 0.500000000 0.351458281
0.543217778 0.000000000 0.183346480
0.457287282 0.000000000 0.811925888
0.042712722 0.500000000 0.688074112
0.956782162 0.500000000 0.316653520
0.865234315 0.000000000 0.544340372
0.136615768 0.000000000 0.449686170
0.363384247 0.500000000 0.050313853
0.634765685 0.500000000 0.955659628
0.153923213 0.000000000 0.945533812
0.346076757 0.500000000 0.554466188