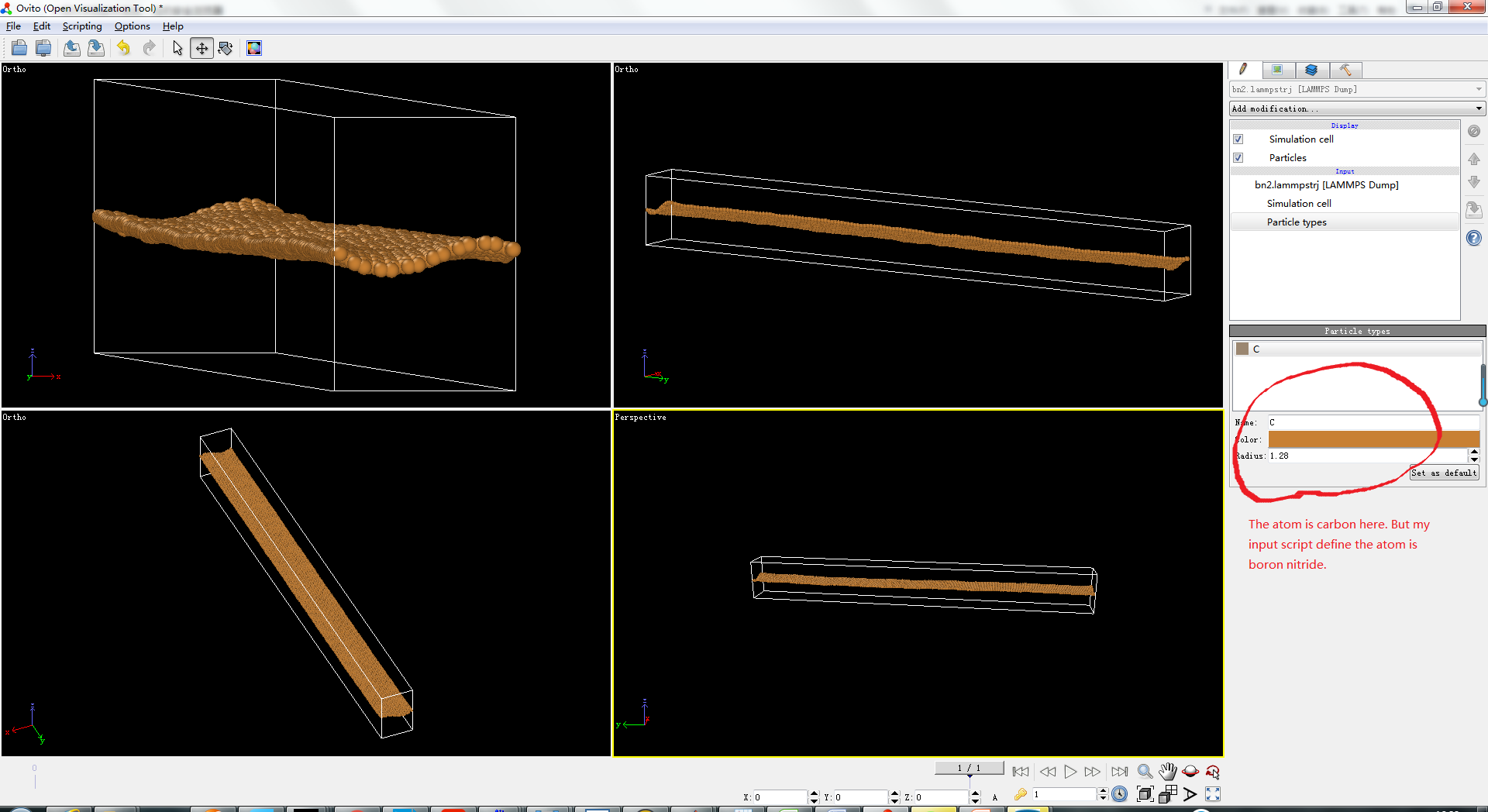
Dear LAMMPS-USERS:
I have been using lammps for 3 months. I want to calculate thermal conductivity of boron nitride by Jund method. Here is my input script. I use BNC.tersoff as my potential. But when i dump the atom information it shows that my atom is carbon ! I also give my ovite picture and dump file in the input script below . I think there must exits some error with my input script. But i can’t find it. Can anyone help me? Thanks a lot.
Imput script:
variable L index 400
variable W index 30
variable H index 45
units metal
processors 1 28 1
dimension 3
boundary p p p
atom_style atomic
#------------------------------Atom Defination------------------------------------------------
lattice custom 1.45 a1 3 0 0 a2 0 1.73205 0.0 a3 0.0 0.0 30 &
basis 0.166667 0.25 0.5 basis 0.333333 0.75 0.5 &
basis 0.666667 0.75 0.5 basis 0.833333 0.25 0.5
region box block 0 $W 0 $L 0 $H units box
create_box 2 box
mass 1 10.811
mass 2 14.0067
create_atoms 2 region box basis 1 2 basis 2 1 basis 3 2 basis 4 1
region hot block INF INF 40 50 INF INF units box
region cold block INF INF 340 350 INF INF units box
#Force-field parameters
#B=1,N=1
pair_style tersoff
pair_coeff * * BNC.tersoff B N
neighbor 1.5 nsq
neigh_modify delay 0 every 10
#---------------------------------Define Setting----------------------------------------------
variable ke equal ke
variable pe equal pe
variable press equal press
variable vol equal vol
variable etotal equal etotal
variable temp equal temp
compute tot_temp all temp
compute myKE all ke/atom
variable temp1 atom c_myKE/0.0001292355
compute hot_temp all temp/region hot
compute cold_temp all temp/region cold
fix INF00 all ave/time 1 10000 10000 c_tot_temp c_hot_temp c_cold_temp v_ke v_pe v_etotal v_press v_vol file INF00_Jund_$L.data
dump 1 all custom 5000 bn1.lammpstrj id element x y z
#---------------------------------------Run---------------------------------------------------
timestep 0.00001
thermo 10000
velocity all create 300 13487594375 mom yes rot yes dist gaussian units box
#-------------------------------------fix npt-------------------------------------------------
fix 1 all npt temp 300 300 0.001 iso 1 1 0.01
run 50000