You’re probably looking for fix ave/atom. The documentation for fix ave/time explicitly states that it only computes averages for global quantities, be they scalars or vectors.
Just a quick query. In LAMMPS documentation, it says:
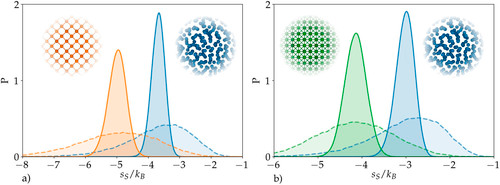
The pair entropy values have units of the Boltzmann constant. They are always negative, and lower values (lower entropy) correspond to more ordered environments.
By “lower values”, does it means “lower absolute values”? If not, then the more negative the entropy per atom is, the more ordered is the environment, correct? However, the second definition does not align with my simulation results as I obtain higher negative values for the crystal and lower negative values for the liquid.