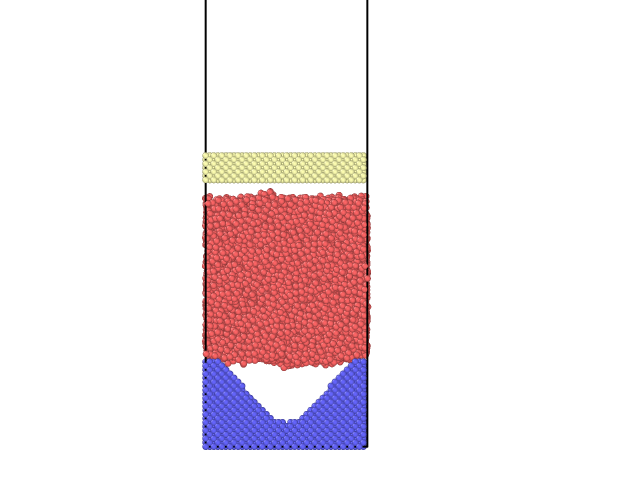
I’ve set up a quasi-two-dimensional sinusoidal surface with a piston on top, and I want to apply pressure by applying a force to the upper atoms, using fix addforce or fix aveforce, but I’m using fix addforce or fix aveforce to tell the upper atoms not to move, right? Why is that? Is there a solution?
My command line is as follows:
variable xlo equal 0
variable xhi equal 71.34
variable ylo equal 0
variable yhi equal 71.34
variable zlo equal 0
variable zhi equal 300
variable sub_lattice equal 3.567
variable n index 4
variable amp equal {n}*{sub_lattice}
variable peri_length equal 71.34
variable off_distance equal 3*{sub_lattice}+{amp}
variable zz internal 0.0
variable v equal "v_zz < (v_amp* cos(v_xx * 2.0*PI/(v_peri_length)) +v_off_distance ) "
variable liquidheight equal 71.34
variable liquidzlo equal {off_distance}+{amp}+3.567
variable liquidzhi equal {liquidzlo}+{liquidheight}
variable upwallzlo equal {liquidzhi}+3.567
variable upwallzhi equal 3*{sub_lattice}+{upwallzlo}
variable upwallfix1 equal {upwallzhi}-{sub_lattice}
variable pressurexlo equal {xhi}/2-5
variable pressurexhi equal {xhi}/2+5
variable pressurezlo equal {liquidzlo}+5
variable pressurezhi equal ${liquidzlo}+15
units metal
dimension 3
atom_style atomic
boundary p p p
neighbor 3 bin
neigh_modify delay 0 every 1 check yes
region box block {xlo} {xhi} {ylo} {yhi} {zlo} {zhi} units box
create_box 3 box
mass 1 18.0154
mass 2 12.0107
mass 3 12.0107
lattice fcc {sub_lattice}
region upwall block {xlo} {xhi} {ylo} {yhi} {upwallzlo} {upwallzhi} units box
create_atoms 3 region upwall
group upwall type 3
lattice fcc {sub_lattice}
create_atoms 2 box var v set x xx set z zz
group downwall type 2
region liquid block {xlo} {xhi} {ylo} {yhi} {liquidzlo} {liquidzhi} units box
create_atoms 1 random 12000 562345 liquid
group liquid type 1
write_data ${n}.data
region liquidpressure block {pressurexlo} {pressurexhi} INF INF {pressurezlo} {pressurezhi} units box
group liquidpressure region liquidpressure
pair_style hybrid sw lj/cut 10.0
pair_coeff * * sw mW.sw mW NULL NULL
pair_coeff 2 2 lj/cut 0.054827 2.629042
pair_coeff 3 3 lj/cut 0.054827 2.629042
pair_coeff 1 2 lj/cut 0.003 2.897291721
pair_coeff 1 3 lj/cut 0.001 2.629042
pair_coeff 2 3 lj/cut 0.001 2.629042
dump 1 all atom 1000 $n101_relax.lammpstrj
fix 1 upwall setforce 0.0 0.0 0.0
fix 2 downwall setforce 0.0 0.0 0.0
min_style cg
minimize 1.0e-12 1.0e-12 10000 10000
unfix 1
compute liquidtemp liquid temp
velocity liquid create 300 3652456 dist gaussian rot yes units box
timestep 0.01
thermo 1000
thermo_modify temp liquidtemp
fix upwall_force upwall aveforce 0 0 -1000 region upwall
fix liquid_nvt liquid nvt temp 300.0 300.0 1
run 10000
Thanks