I have run it for 1ns for zero pressure at 300K . But my lattice size keeps increasing abruptly. Attached is my script and last lines of the log file:
Script:
Your neighbor list updates are likely too infrequent and thus you may get inconsistent computation of interactions due to missing neighbors when using an outdated neighbor list.
default was 1 step for every, and actually you are right I got smaller lattice but still there was an increase. Can that be because I started at a lattice size 5.431 which was already given as the equib. value for Si in the literature. ( I just wanted to see if it is working fine with the code)
If you look at the output you can see that you have a neighbor list update every 24 steps on average, but that almost half of them were “dangerous”. so delay 10 is probably ok, but every should be shorter, 5 or 2. there isn’t much speedup to be gained after that. by using delay 10 you already reduce the cost of updating the neighbor list to 1/10th. reducing the remaining 10% cost to 5% is not going to save you much but very much increases the risk of faulty neighbor lists. It usually is not a problem for a solid bulk system to have a few dangerous builds. But they should not be at the level of what you have.
You have to look for the lattice parameter that is consistent with the specific potential parameters that you are using. In simulations there is not the lattice parameter and it rarely is identical to the experimental value.
Not to mention that you have to compare to a value to the same thermodynamic conditions.
In addition, you have to check whether the residual stress is isotropic, when using fix npt with the “iso” keyword you enforce that. While looking at the fix npt parameters I also notice that your barostat relaxation time is shorter than your thermostat relaxation time. That is very unusual. Typical would be to make that larger. This parameter can be critical for lattice relaxation and may be a contributor to unwanted fluctuations if too small.
To get a reliable estimate for lx you must use averaging and you need to choose settings for fix npt that keep fluctuations low. Since it is easier to expand than to compress a system, large fluctuations usually are not following a normal distribution so the average can change significantly if the fluctuations are large. Sometimes it helps to use the (empirical and thus unphysical) drag parameter to reduce fluctuations in solids. Due to their incompressible nature, fluctuations are naturally larger than in liquids (and typical recommendations for settings you find for fix npt tend to be for liquids, not solids).
Thank you Alex for your time. About the thermostat and barostat relaxation times, actually I mistakenly put them like that, they should be flipped. It looks it helped, I am getting better numbers closer to the value.
Thank you very much.
Update: It looks like it worked better, I ran the sim for 2ns, started with 5.5A lattice size and ended at about 4.905. So it shrank little bit.
1990000 -17690.83 49.076626 -202.99923 300.16948
1991000 -17693.607 48.974005 679.86826 302.55388
1992000 -17689.71 49.109225 -1250.1539 301.37983
1993000 -17693.507 49.015775 343.09167 304.5067
1994000 -17689.084 49.024077 215.16569 299.63423
1995000 -17690.391 49.117265 -689.96654 304.31459
1996000 -17694.278 48.976194 835.25828 299.61107
1997000 -17693.286 49.009782 165.60848 301.53058
1998000 -17690.812 49.055964 367.79 298.90226
1999000 -17692.112 48.9973 -102.91499 292.73121
2000000 -17692.848 49.067537 -445.41547 306.34811
My question is the Pressure here which is not stabilizing around 0 same for Temperature which is not quite well stable at 300K. What would you suggest?
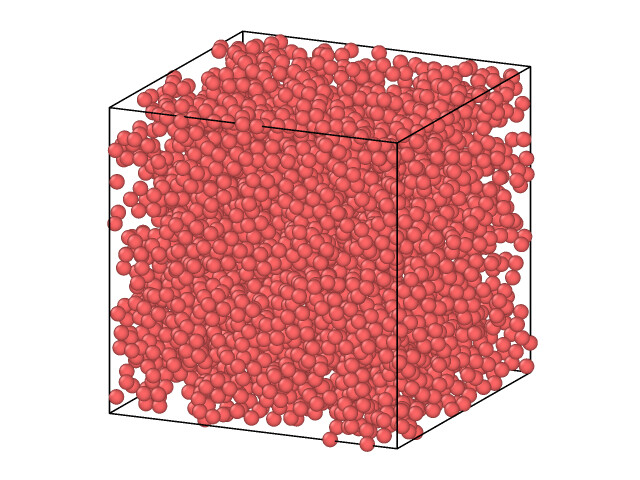
a lattice size 5.5 initially with 10 lattice on each direction generated 4000 atoms, but this is what I got after 2ns with a porous structure for Si. Am I missing sth?
This has been discussed many times previously. Please check the mailing list archives. I also mentioned it in a previous response.
Bottom line: pressure fluctuates and by a much larger amount than temperature; specifically for a system that is not very compressible. Please consult a statistical thermodynamics text book where you can learn that correspondence to bulk properties from atomic scale properties usually requires averaging and for a system in equilibrium the average over time is equivalent to the average over space.
Thank you for your time. I read your previous posts Just wanted to ask to make sure it is not sth due to the input script.
Coming back to the last part, sorry for the confusion. I just posted the image showing the 5x5x5 nm box with 4000 Si atoms after 2ns of NPT run at 300K. I would not expect to have this porous structure. I did not enter the number of atoms, 4000 is what was generated when I entered the lattice size, mass with the Si.tersoff potential.
You asked to generate an FCC lattice, but crystalline Si is not FCC, so it does not want to be in your starting configuration, but at 300K there is not much that can happen.
Yes Alex, Thank you. I never looked at that part of the code. I missed it. Now it runs better, with the lattice parameter running around what has been reported in the lit.
 Just wanted to ask to make sure it is not sth due to the input script.
Just wanted to ask to make sure it is not sth due to the input script.