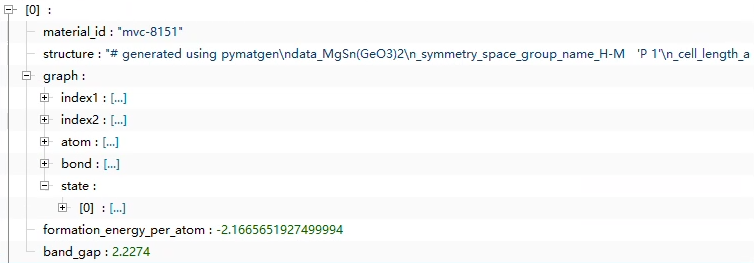
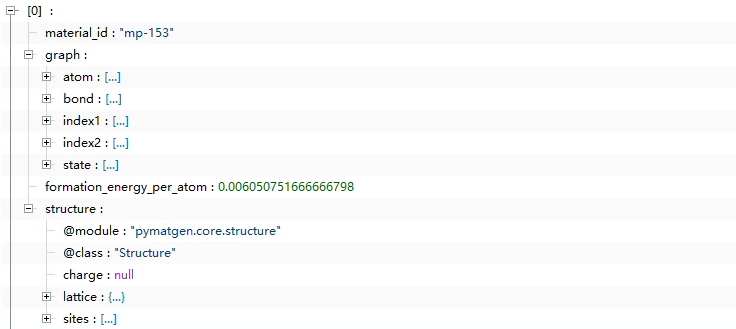
I used the megnet project on GitHub to build the database by myself, using the method of cutoff = 5 in convert as mentioned by the project, but the structure I came up with is not the same as his. I want to ask the builder of the dataset to help me
! [image | 690x240] (upload://rTF4OSvvOIAoSP2ObS35DnYSmiH)
! [image | 690x308] (upload://oe3Uz8ifoiMzBwODeFbKW3m9jQf)
The first image is from the official MP - 2018.6.1 Graphs of materials project
The second picture is the result of my attempt to reproduce it according to the megnet project. Here is my code. I would like to ask for your help. This is very important to me because I have not found the complete sample code for processing data and training code on GitHub
Import warnings
Warnings.filterwarnings (‘ignore’)
From megnet.data graph import GaussianDistance
From megnet.data crystal import CrystalGraph
From megnet.models import MEGNetModel
Import pymatgen as pm
From pymatgen.ext.matproj import MPRester
Import numpy as np
Import json
#Initialize MPRester,
api_key = "
MPR = MPRester (api_key)
material_ids = [
Acquire multiple crystal structures
Structures = [mpr. get_structure_by_material_id (mid) for mid in material_ids]
#Initialize the list used to store all crystal structure data
all_data =
For i, structures in enumerate (structures):
#Use the convert method to create a CrystalGraph instance and set the cutoff
crystal_graph = CrystalGraph (cutoff = 5.0)
Graph = crystal_graph convert (structure)
#Extract node features
Atom = graph.get (‘atom’, )
#Extract edge features
Bond = graph.get (‘bond’, )
#Extract Atomic Index 1
Index1 = graph.get (‘index1’, )
#Extract Atomic Index 2
Index2 = graph.get (‘index2’, )
#Assuming there is no specific state information here, state is set to None
State = graph.get (‘state’, )
Acquire the formation energy
formation_energy_per_atom = mpr.get_data (material_ids [i], prop = ‘formation_energy_per_atom’) [0] [‘formation_energy_per_atom’]
#Convert data that may be of type numpy.ndarray to a list
If isinstance (atom, np.ndarray):
Atom = atom.tolist ()
If isinstance (bond, np.ndarray):
Bond = bond.tolist ()
If isinstance (index1, np.ndarray):
Index1 = index1.tolist ()
If isinstance (index2, np.ndarray):
Index2 = index2.tolist ()
If isinstance (state, np.ndarray):
State = state.tolist ()
Organize the information related to the graph structure into a dictionary
graph_info = {
'Atom ': atom,
'Bond ': bond,
'Index1 ': index1,
'Index2 ': index2,
'State ': state
}
Build a data dictionary of the current crystal structure
Data = {
'material_id ': material_ids [i],
'Structure ': structure.as_dict ()
'Graph ': graph_info,
'formation_energy_per_atom ': formation_energy_per_atom,
}
all_data append (data)
Save all data as a JSON file
With open (‘crystal_graph_dataset json’, ‘w’) as f:
JSON.dump (all_data, f, indent = 4)