Dear all,
I use
fix bottomf wall setforce 0.0 0.0 0.0
to hold graphene sheet static and I use sw potential setting as
C C C 0.25956 2.3925 1.8 0 1.2 -0.333333333 0 0 4 0 0
when I
compute pe_carbon_atom wall pe/atom
compute pe_carbon wall reduce sum c_pe_carbon_atom
thermo_style custom step c_pe_carbon
I get c_pe_carbon output -nan, I am just wondering that shouldn‘t static atoms should also have potentials energy?
Yes, what you do is consistent with the documentation and the semantics of what your calculation is doing. With the fix setforce command — LAMMPS documentation you are erasing all previously computed forces and then replace them with new values. That does not change the computation and thus also not the computed potential energy. If you get “nan” values, that usually means you have overlapping atoms.
That should be investigated and resolved, as that will result in unexpected behavior of those atoms with the rest of the system (at every overlap point your interaction will be doubled).
I examined my carbon atoms and found no overlap. As all my atoms are static, it’s very easy to check if there is overlap. I am now really confused about the reason why would this nan appear. Do I need to upload my full script?
Please do not upload the full script. Instead try to create an input deck that has as few commands and creates as few atoms as possible yet still produces the NaNs.
restart.eq (4.8 MB)
system.in (1.8 KB)
system.sw (901 Bytes)
here is simplest version of my script to reproduce the problem. I am using lammps16Aug18 mpirun -np 6. This problem also arises when I use lammps7Aug19. Actually there are two problems here
- in thermo output, c_pe_carbon is NAN as I said upwards
- after 20596 step, all my water atoms info output NAN
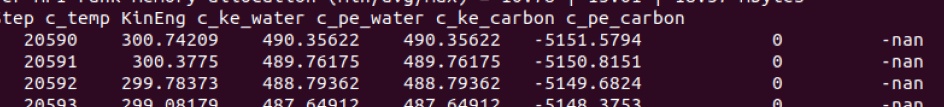
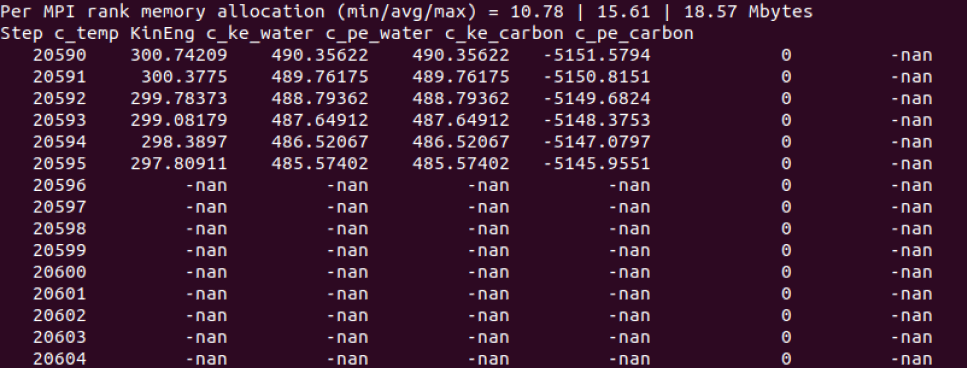
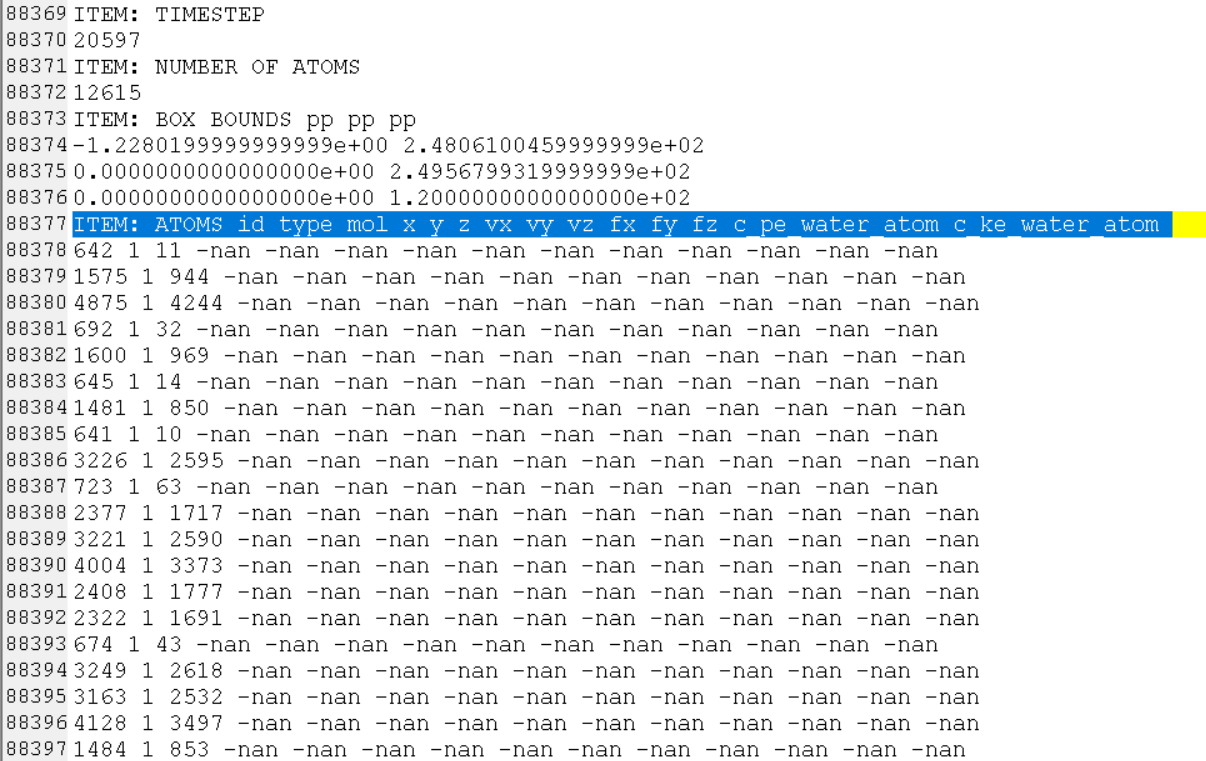
I examined the trj and found at timestep 20596, all my velocity output NAN and there is one atom whose force also output NAN, atom 642

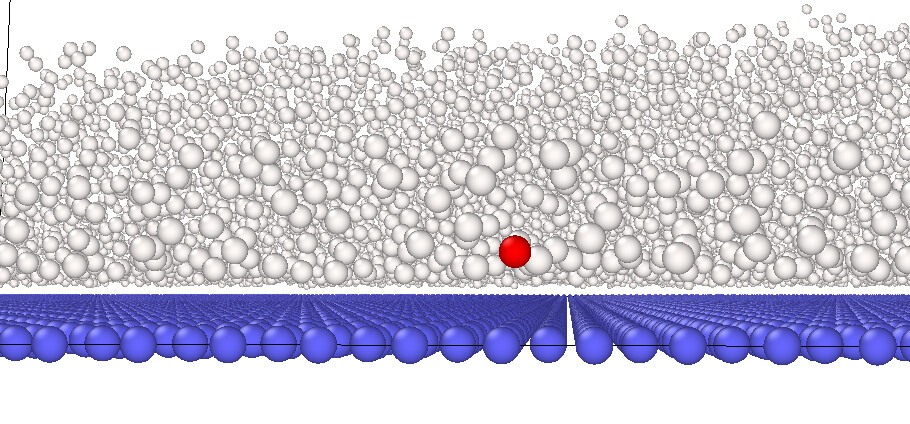
but visualization shows that 642 atom is actually not overlapping with any other atoms (red atom in the following pic is atom 642)

I have uploaded a simple script to produce the problem. actually I have run this script more than 10 times at different initial velocity configurations, the NAN of the pe_carbon always persists and the NAN of atom coords comes sooner or later and the simulation eclapsed at all events. here is my scipt to get this restart file.system.in (2.2 KB)
system.sw (901 Bytes)
WaterCarbonSystem.data (1.4 MB)
When I add the line
delete_atoms overlap 0.1 all all
To your input right before the first run command, I get the following report:
System init for delete_atoms ...
Neighbor list info ...
update every 1 steps, delay 1 steps, check yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 7.76
ghost atom cutoff = 7.76
binsize = 3.88, bins = 65 65 31
2 neighbor lists, perpetual/occasional/extra = 1 1 0
(1) command delete_atoms, occasional, copy from (2)
attributes: full, newton on
pair build: copy
stencil: none
bin: none
(2) pair sw, perpetual
attributes: full, newton on
pair build: full/bin
stencil: full/bin/3d
bin: standard
WARNING: Ignoring 'compress yes' for molecular system (src/delete_atoms.cpp:126)
Deleted 102 atoms, new total = 36585
This means that LAMMPS has detected that there are 102 atoms with a distance closer than 0.1 angstrom. As I already mentioned that is likely due to periodic boundaries and that means that either you have not created the positions in the data file correctly or given incorrect box dimensions (e.g. by using min/max coordinates without leaving room for the atom diameters themselves since the min/max determination is for point particles without a diameter).
When the run continues after the delete, the NaNs are gone:
Step c_temp KinEng c_ke_water c_pe_water c_ke_carbon c_pe_carbon
0 100 163.04875 163.04875 -2843.029 0 -35.654049
1 Like
Thanks a lot, it really helps me!