Hi everyone,
I want to determine the size of the bulk cell to be used. I check different bulk cell size.
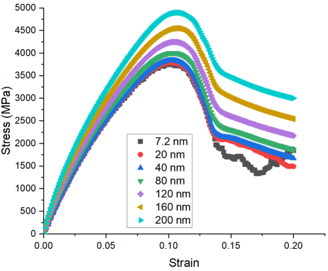
When doing the compression of copper single crystals, except for the size of the bulk cell I used, the other conditions used are the same (periodic boundary conditions are applied). Why do the resulting Young’s modulus and yield strength increase as the bulk cell size increases? Why am I not seeing convergence? Can anyone give me some advice about this? Thank you in advance!
The following are the script I used:
------------------------ INITIALIZATION ----------------------------
units metal
dimension 3
boundary p p p
atom_style atomic
variable latparam equal 3.597
----------------------- ATOM DEFINITION ----------------------------
lattice fcc {latparam}
region whole block 0 55 0 55 0 55
create_box 1 whole
region upper block INF INF INF INF INF INF units box
lattice fcc {latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 region upper
------------------------ FORCE FIELDS ------------------------------
pair_style eam/alloy
pair_coeff * * Cu01.eam.alloy Cu
------------------------- SETTINGS ---------------------------------
compute csym all centro/atom fcc
compute peratom all pe/atom
######################################
EQUILIBRATION
reset_timestep 0
timestep 0.001
velocity all create 300 12345 mom yes rot no
fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1
Set thermo output
thermo 1000
thermo_style custom step pxx pyy pzz pe temp lx ly lz press
run 300000
unfix 1
Store final cell length for strain calculations
variable tmp equal “lx”
variable L0 equal {tmp}
print "Initial Length, L0: {L0}"
######################################
DEFORMATION
reset_timestep 0
fix 1 all npt temp 300 300 1 y 0 0 1 z 0 0 1 drag 1
variable srate equal 1.0e10
variable srate1 equal “-v_srate / 1.0e12”
fix 2 all deform 1 x erate ${srate1} units box remap x
variable strain equal “(lx - v_L0)/v_L0”
variable p1 equal “v_strain”
variable p2 equal “-pxx/10000”
variable p3 equal “-pyy/10000”
variable p4 equal “-pzz/10000”
fix def1 all print 100 “{p1} {p2} {p3} {p4}” file Cu_comp_100.def1.txt screen no
Use cfg for AtomEye
dump 1 all cfg 100 dump.comp_*.cfg mass type xs ys zs c_csym c_peratom fx fy fz
dump_modify 1 element Cu
thermo 1000
thermo_style custom step v_strain temp v_p2 v_p3 v_p4 ke pe press
run 20000