Hey there,
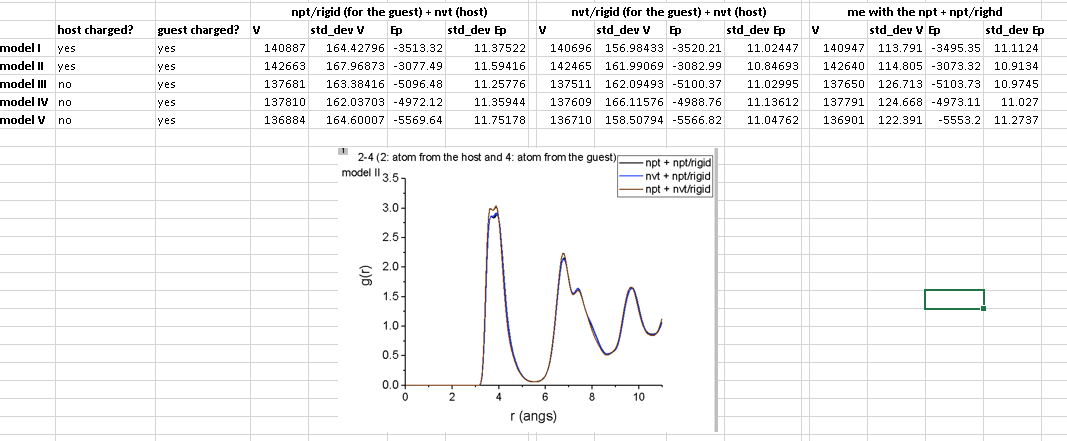
I recently found myself doing some classical MD simulations of a metal-organic framework (host) loaded with a given amount of N2 molecules (guest). My goal was to simulate the dynamics in the NPT ensemble and evaluate the resulting structure of my system once the equilibrium state had been reached. My initial microstate contained information on position of the framework (based on its crystal structure at ambient conditions) and a given fixed amount of N2 molecules (which were in somewhat “random positions”). I wanted to simulate a point of the experimental isotherm, meaning that my goal was to equilibrate the system containing that given, fixed amount of N2 molecules at the pressure which is observed to be the corresponding equilibrium one at that loading.
In short terms, the potential I was using for my N2 molecules required them to be rigid. At the same time, I wanted the atoms of my framework to move relative to one another. Since I wanted to simulate the dynamics in the NPT ensemble, the first idea that had come to mind was to define a fix npt/rigid to set up the dynamics of the guest and a fix npt to set up the dynamics of my host (example on how this would look like below). However, in the manual there is a mention about how we should not set up a ‘hybrid’ simulation where a part of the system evolves according to npt/rigid and another part with npt, the reason being “because there can only be one fix which monitors the global pressure and changes the simulation box dimensions.”
fix 16 guest rigid/npt/small molecule temp 77 77 100.0 aniso 0.0538 0.0538 1000.0
fix 1 host npt temp 77 77 100.0 aniso 0.0538 0.0538 1000.0
run 4000000
unfix 1
unfix 16
I am aware of the ways I could do my simulation (using for example nvt/rigid for the dynamics of my molecules), but I kind of just wanted to check if I understood the issue with using npt/rigid for one part of the system and npt for the other. As I am not defining some weird partial pressure computation for any of the fixes by means of a fix_modify, the pressure compute inherent to both fixes above would cherish the contributions of all the atoms composing the system. However, the standard of a fix npt/rigid is to decrease the number of degrees of freedom so that each rigid body is accounted as one entity in the computation of pressure. At the same time, the same would not done for the fix npt in the scope of its calculation of pressure (meaning it would account each atom of my rigid molecules of the guest as one atom), right? This is the (only) issue, right? Because be it in the equations shown in Shinoda et al paper (mentioned in the fix npt page of LAMMPS manual) or the basic Nose-Hoover approach to treat the dynamics of the system in the NPT ensemble, I would suppose that as long as I am using the same Text, Pext, Tdamp, Pdamp, I would have the value of my “ficticious variables” as well as their time evolution be the same in the context of both my fixes. Same would therefore hold for the predicted change in volume at each timestep.
PS: By “ficticious variables” I mean those invented to mimic the interaction with the surroundings in the context of simulating the system in an NPT ensemble. For example, looking at the equations for the basic Nose-Hoover approach shown below, it would be zêta and êta.
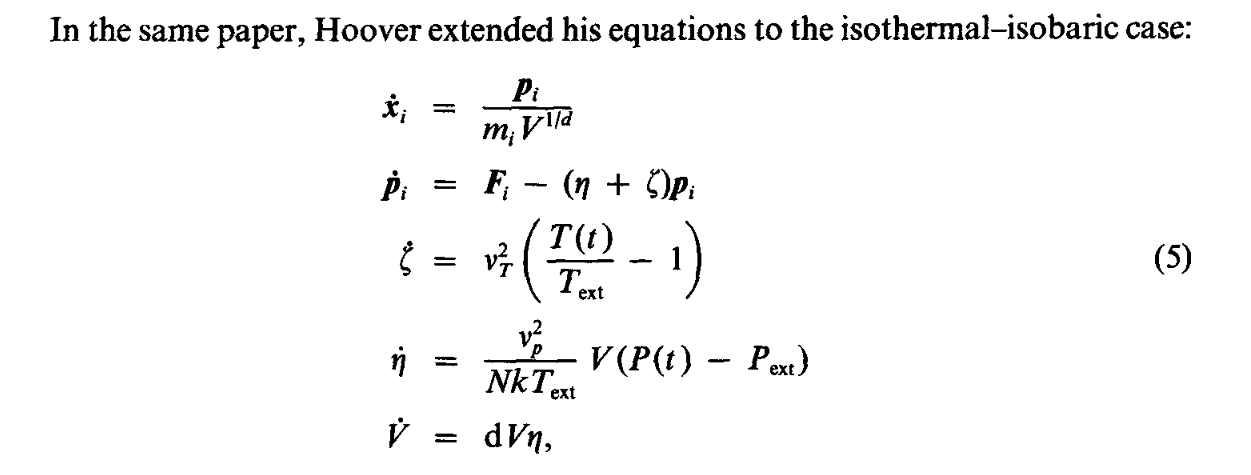
Source: Melchionna, S., Ciccotti, G., Holian, B. L. 1993, Molec. Phys., 78, 533. ( https://doi.org/10.1080/00268979300100371)
Thanks for the help,
Best,
Cecilia