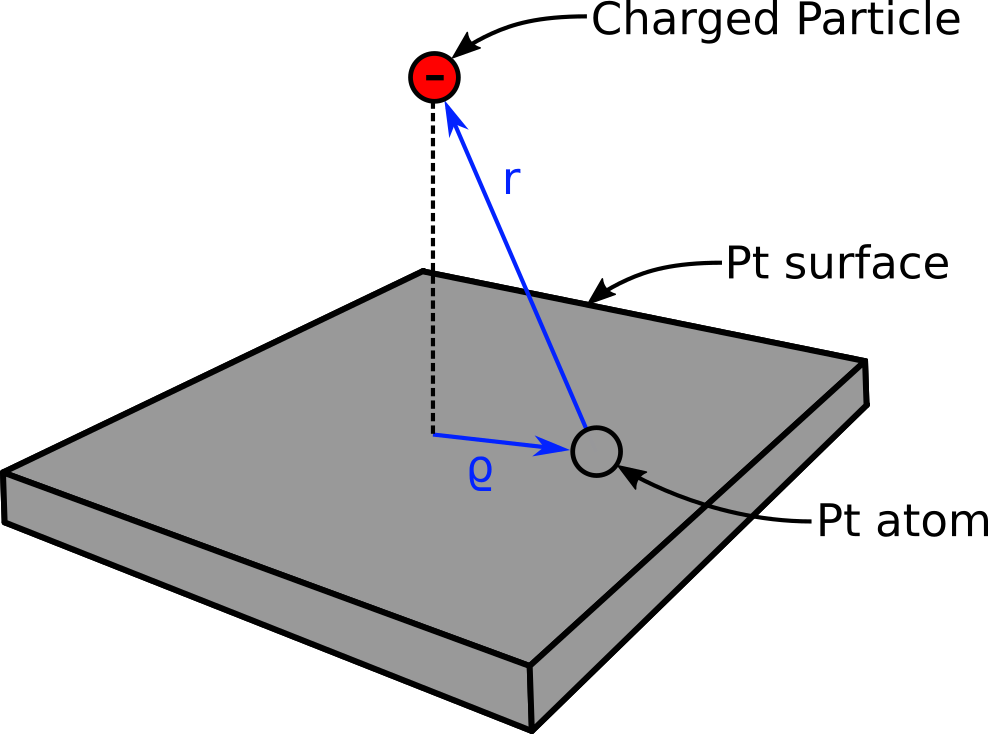
I’d like to implement the Spohr potential in LAMMPS. If you’re unfamiliar with the Spohr potential, it’s essentially a Morse potential with an additional term to account for charges induced on a metal surface. I’ve included a picture to make things a little clearer:
The potential is dependent on the distance between the charged particle and the surface, and the projection of this distance vector onto the surface.
I’m trying to find a good starting point for writing this. I was thinking of just modifying the pair_morse.cpp to include the image charge term. It seems like it would involve just using the x and y components of the distance vector to calculate the projection. I was also reading another post a while ago (which I can’t seem to find now), where Axel suggested modifying fix_rigid_small.cpp to achieve the same result. However, fix_rigid_small.cpp is quite a bit more complicated than pair_morse.cpp.
Any thoughts on using either fix_rigid_small.cpp or pair_morse.cpp, or ideas for other fixes/pairs to start out with would be greatly appreciated.
This was likely in reference to the “image charge model” which the paper you are quoting does not implement. In the image charge model, you define a “mirror” plane (that is material specific) and then assume the metal is perfectly polarizable, so that all charges are mirrored (also in sign) at the image plane and then those point charges are included in the coulomb interactions.
What you have here is simpler. Starting from the Morse (or any other simple pairwise potential) is a good starting point. I would suggest the following strategy:
in the Pair::settings() function you need to process additional parameters and parse/store them. you need: the atom type of the water oxygen atoms, the atom type of the water hydrogen atoms, the atom type of the Pt atoms, the orientation of the Pt surface (or you can hardcode it to be in the xy-plane for simplicity).
in the Pair::coeff() function I would just accept and parse the cutoff or make it not parse any arguments and require a global cutoff only
in the Pair::compute() you have to check the atom types and then branch into either O-Pt or H-Pt (or Pt-O and Pt-H) interactions and skip everything else. That means you do the water-water with the existing potentials using pair style lj/cut/coul/long in combination with your custom pair style via pair style hybrid. You have to make certain that your Pt atoms have no charge. The force and energy computation should be straightforward, when you can assume that the Pt surface is aligned.
there is minor work needed for restarting and bookkeeping etc.