For the latest ovito (actually also for the recent ovito versions), when I export a file to lammps data,
the file will look like
# LAMMPS data file written by OVITO Pro 3.14.1
16573 atoms
4 atom types
60.9763723175 0.0 0.0 avec
0.0 61.0008627877 0.0 bvec
0.0 0.0 59.9828566258 cvec
0.9616840196 0.9485835072 80.0149758206 abc origin
However, it should be like
# LAMMPS data file via write_data, version 29 Aug 2024, timestep = 1470000, units = metal
42000 atoms
4 atom types
0.9477354616024852 61.952264538398126 xlo xhi
0.9477354616024852 61.952264538398126 ylo yhi
2.371598754472661 155.02840124552972 zlo zhi
This is not a bug. Both formats are valid LAMMPS data files, as documented in the LAMMPS manual.
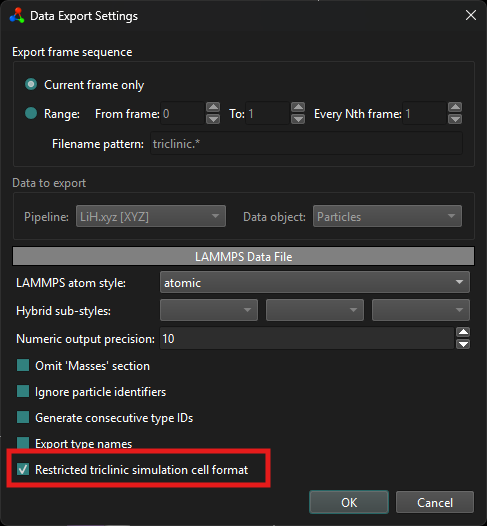
The first output format you are seeing is the restricted triclinic simulation cell format. You can select it using this checkbox in the data export settings:
I checked your files and even the unit/super cells are not recognized as HCP phase by OVITO. What c/a ratio does your material have? OVITO usually expects a c/a ratio reasonably close to the ideal 1.63. Are you sure you handled the rotation and cutting of your unit cell correctly?