hello, everyone:
I am reading the fix evaporate command in lammps doc.
This command remove M atoms from the simulation every N steps, but there is no information for the energy.
my question is: if this fix delete the M atoms, does the (potential) energy from these atoms will be delete?
Hi,
Why not make a quick test with a simple input where you print the potential energy of a simple system undergoing evaporation from fix evaporate?
Simon
1 Like
Yes, you are right, I will run a test and update the results here
1 Like
This is a question of fundamental physics and completely independent of LAMMPS. The potential energy of a system is a state function, i.e. it is fully described by the current state and it is irrelevant how you got there.
Dear Axel,
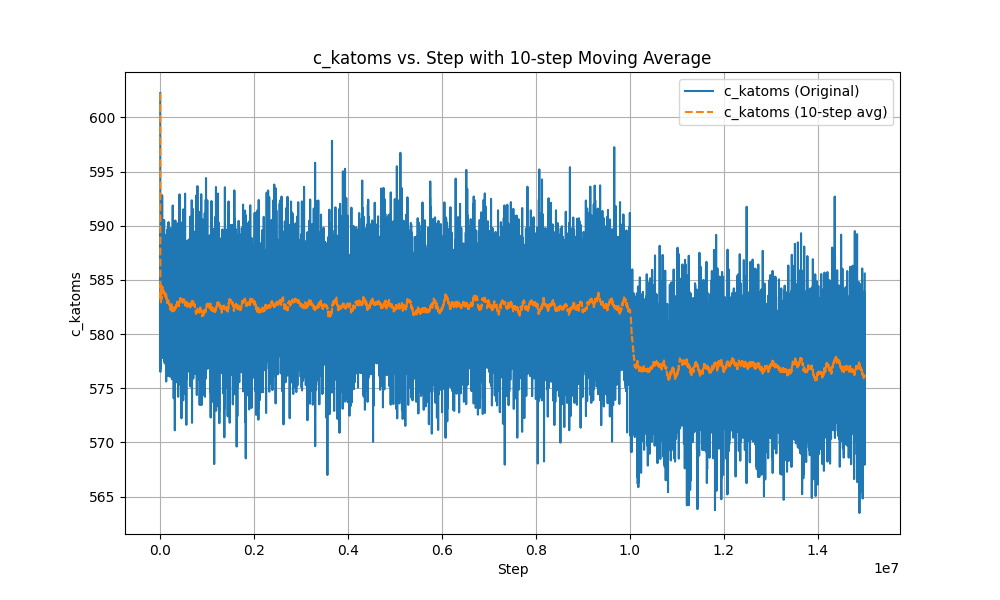
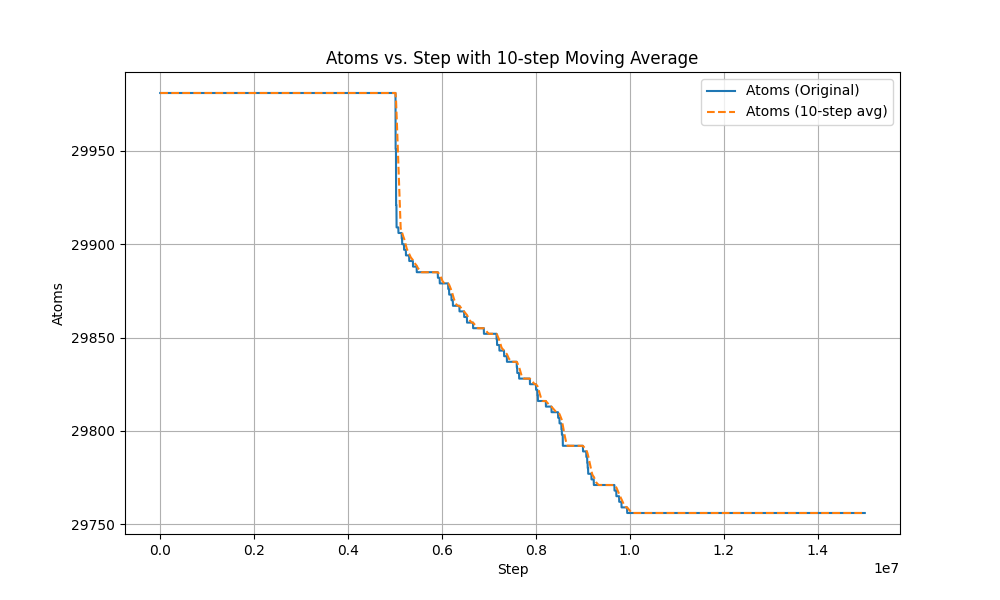
I conducted a simulation of water evaporation and obtained some thermodynamic quantities of the system. In the first 5 million steps of the simulation, I did not use the ‘fix evaporate’ command. In the subsequent 5 million steps, I employed the ‘fix evaporate’ command, and in the final 5 million steps, I turned off this command. From the results, it was expected that the total kinetic energy of the system would decrease between the 5 millionth and 10 millionth steps due to the continuous removal of water molecules. However, based on the output, there was no observed decrease in total kinetic energy during the ‘fix evaporate’ process. Surprisingly, when I turned off this command at 15 million steps, the kinetic energy suddenly dropped and remained stable. Therefore, I suspect there might be some bugs in the simulation?
There is not enough information and explanation here to make any meaningful assessment.
in.evap (1.7 KB)
Sorry, here is my input
This is rather useless, specifically since it is missing the data file. Furthermore, if you want to have somebody have a closer look, it has to be possible to reproduce the effect you are seeing with only a small number of processors and rather quickly. Thus you need to produce a small test system and reduce the number of timesteps between the different stages.
The plot you are providing is rather useless. If you want to observe whether atoms are evaporated, you need to monitor (and thus plot) the number of atoms in the system.
Furthermore, you need to visualize your system and see if there is a significant change happening during the “drop” in kinetic energy. Does it affect the temperature/pressure or potential energy?
You don’t explain which LAMMPS version you are using, what platform you are running on, what your command line is and so on.
Just dumping the input on people and thinking this explains everything is overestimating the capabilities of people answering here and somewhat rude. People are the more likely to help you, if they see that you are making an effort. At the moment this is not the case. Please see the forum guidelines for some more explanations and examples.
Thank you, Mr. Axel. I just celebrated the Spring Festival and saw this reply from you. I’m really sorry.
The system is composed of Cu and water interface as shown in the figure.
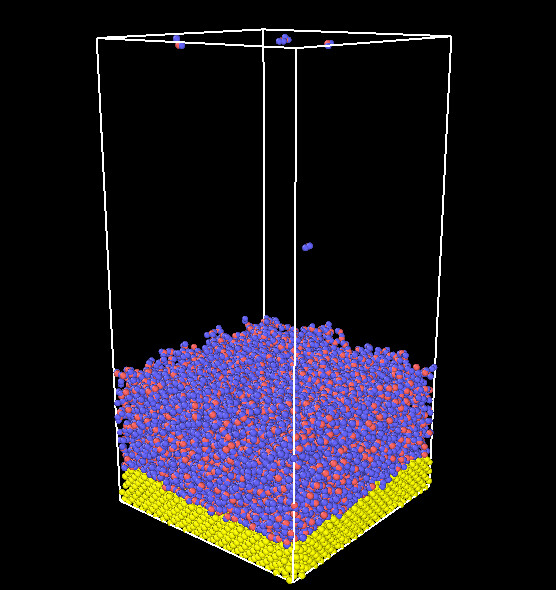
When the water is evaporated in the bulk, it floats to the top of the box. when a molecule is in the evap_space, it will be deleted. Through the fix evaporate command, I did observe a decrease in the number of atoms, but correspondingly, the energy of the system should decrease as the number of atoms decreases, but the simulation results are not like this.the atom-steps is shown in the figure.
the tempreature plot is shown in this figure.

The LAMMPS version is 2 Aug 2023, running mpirun with 64 cores on CentOS system.
I haven’t seen the source code for fix evaporate, so I didn’t know what happened in this process.