I am posting anew because I have done some additional work to pin down this overprediction of density by LAMMPS. Many details can be found in the thread above.
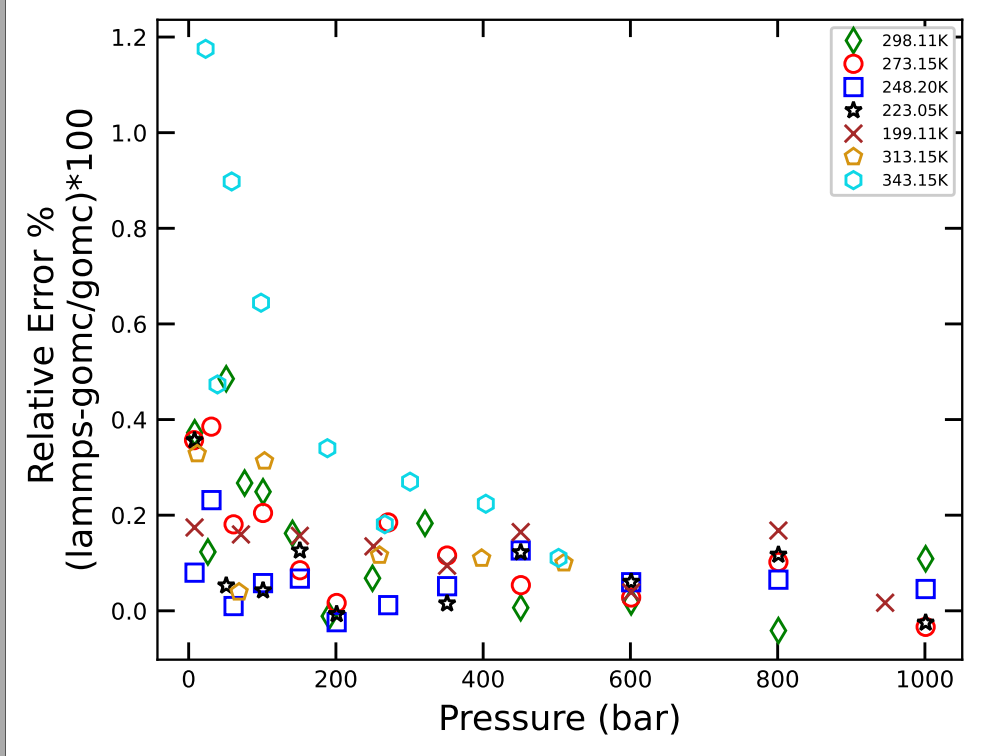
I have not yet been able to solve this overprediction of density by LAMMPS. I am running a NPT simulation, and for a very particular state point (T = 343.15K and P=23.03 bars), I get density difference > 1%.
I have checked the same state-point with following MC / MD engines and all (except LAMMPS give me density to be around 1007 kg/m3. LAMMPS give me 1019 kg/m3.
GOMC (MC) : 1007
GROMACS (MD) : 1008
pyMCMD (cyclic switching between GOMC & NAMD) : 1007
LAMMPS : 1019
In my opinion, I have pretty much exhausted all possibilities to explore and pin down the reason for this difference. I have also tried different equilibration protocols as:
same protocal as above with addition of Berendsen+nve before using Nose-Hoover Barostat
minimize > langevin_thermostat+nve > nvt > npt
But there is no appreciable difference and equilibrated density always is 1019.
One other important point is that this density difference is pronounced and very systemic for one particular temperature and pressure. I have explored many other temperature / pressure regimes and I am including the graph for further elaboration.
Well, clearly the central C-C bond vibrations are to blame. This is backed up by the trend that the discrepancy increases with higher temperature and lower pressure: at lower pressures intramolecular forces dominate intermolecular forces, and at higher temperatures vibrational modes have more energy.
I mean, sure, you can do that (do remember though that when a bond is too stiff the integration timestep needs to be reduced to properly integrate its motion).
I’m just deeply curious as to why you’re so concerned about a 1% density difference – and not even between experimental data and your calculations, but between two different software packages that are effectively implementing two different force fields. (A force field includes all parameters needed to calculate the forces, and thus a bond with effectively infinite rigidity is different from a bond with a high but finite bond constant). “High precision” and “molecular dynamics” are not words that usually go together.
Actually, I want to first validate the LAMMPS implementation of FF. So that I can go on to do more interesting things with this compound in LAMMPS. But for that, I need to get densities in acceptable range. I am worried that there is some small detail that I am doing wrong in LAMMPS.
I understand that FF are same for both MC & MD (besides this bond issue as GOMC does not sample bond).
If you look at the systemic increasing trend at high temp (as pressure is decreased) which again points out something systemic and not statistical scatter.
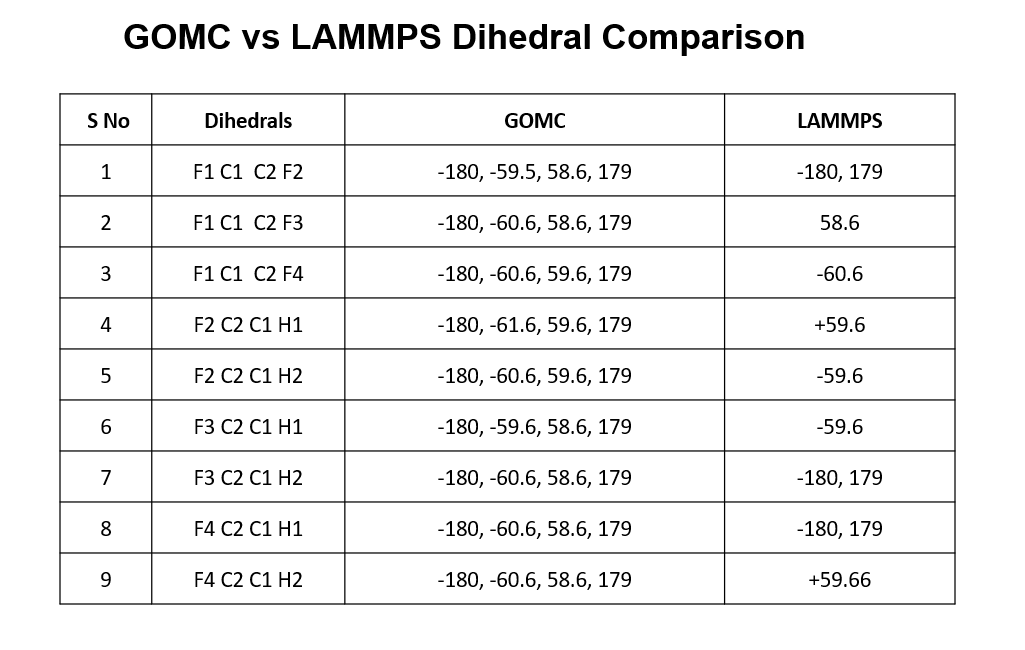
Since you have already mentioned something’s going on with intramolecular forces, my mind is kind of stuck on dihedrals. I used vmd tcl scripting (.dcd) to see what dihedrals are exactly being sampled in both GOMC and LAMMPS. There are 02 types of dihedrals and total 9 dihedrals. I found out that in GOMC, all 9 dihedrals sample correctly (±60, ±180) but lammps behavior is strange. My results are enclosed in the image below. I am wondering if this could be the reason of this behavior. And if so, how to get LAMMPS sample all proper dihedrals.
Thank you again for taking the time out to read this thread and provide your valuable suggestions. I really appreciate it.
No, they are not the same. They are almost the same, which is why your results are almost consistent.
I hate to be harsh, but you have spent three months on this issue simulating your system in four different softwares at about 40 different state points, if the graph in your first post is correct. At 20 ns per trajectory (which I’d start with to test convergence) that’s a few microseconds’ worth of simulation time. Are you sure this has been a good use of your computer time? Since you know for sure that GROMACS reproduces the GOMC results, why not just use GROMACS?
Unless you are explicitly writing a computational paper comparing different softwares, you should simply pick the package that gives you the most consistent results and go with it. I’m not sure what exactly you’re thinking of doing so that you’re so focused on getting the LAMMPS force field working, but you really should check if you can do the same thing in GROMACS since it can rigidify that C-C bond and LAMMPS can’t.
Regarding the dihedrals, the most likely explanation is that Monte Carlo, if the steps are big enough, can “jump” barriers in a way that molecular dynamics can’t, since an MC step is accepted just based on the energy difference between initial and final state while in MD a transition must get over the activation barrier.
I know its been quite some computer and clock time!
Reason for exploring LAMMPS only is to work on different kinds of transport properties, S/L equib, VLE and so on. I am a bit more familiar with lammps implementation and that’s why am trying to get this working.
I will also look to get it working in GROMACS now.
This is so much of a waste, because you have picked a very complex and indirect parameter in order to validate the implementation of a force field. What would have been far more effective (and still is, regarless of whether you are using LAMMPS or not) is to compare the force and energy computed for individual parts of the force field. By that I mean that you set up a calculation with just a pair of atoms (or more for bonds, angles, dihedrals) at various distances (or geometries) and get the energies and forces with all other force field components turned off; and where no forces are computed, you can compare \frac{\Delta U}{\Delta r}. LAMMPS even has a fix to automate that: fix numdiff command — LAMMPS documentation. If you have validated each component of the force field, you need to validate how exclusions, cutoffs and so on are computed and how different codes stack up against each other. If done with the proper care, you should be able to tell whether there are differences between the different implementations at the very basic level. If there are none, you have to gradually check each and every component that impacts the propagation of atoms. If the implementations are all correct, they should yield the same results, not just for density, but if you reduce the number of degrees of freedom, it should be possible somehow to identify where there is a difference.
Nobody deliberately writes wrong software, but there are often very subtle differences in the choices that people make in their implementation that make inputs that, do look the same and have the same settings, still behave differently, if only in subtle ways. A common issue would be how exclusions are handled. There may be subtle differences there, and also - for example - how exclusions are computed in the presence of long-range electrostatics. I recall that we had some very subtle changes done on the LAMMPS implementation for how to apply exclusions consistently in that case a long time ago. If you are not so familiar, you may not have chosen settings that are not 100% equivalent. If this you should create a test case comparing cutoff coulomb instead of PPPM or Ewald or PME.