I have calculated densities for R134 at various temperatures and pressures. For MonteCarlo, I have used GOMC software. For MD, I am using LAMMPS and comparing the two. LAMMPS is systemically overpredicting density over all state points. Percentage error is low at lower temperatures, but it the highest for highest temperature & lowest pressure.
I am using same forcefields and same parameters like cutoff, electrostatics etc. For the worst state points, largest error in density is up to 1.2%.
I did minimization and equilibration in nve/limit > nve > nvt (50 picosecond) > nvt (2 ns). I am also attaching my lammps script for final npt production run.
Any ideas of why this higher density by LAMMPS.
PS : I also did Single Point Energy calculation (same snapshot) for both MC & Lammps. Percentage differences in LJ, tail corrections, electrostatic are to the order of 1E-3 percent. However, pressure difference is 13 percent which is very high. Probably that’s why the difference in averaged density using NPT?
Since I cannot upload attachment as I am new, I am putting my NPT production script here:
units real
dimension 3
newton on
atom_style full
variable V equal vol
variable d equal density
variable t equal temp
variable pres equal press
variable startstep equal step
variable evdwl equal evdwl
variable ecoul equal ecoul
boundary p p p
pair_style lj/cut/coul/long 14.0
pair_modify mix arithmetic shift no tail yes
bond_style harmonic
angle_style harmonic
dihedral_style opls
read_restart minimized-0.in
kspace_style pppm 1.0e-5
neighbor 6.0 bin
variable T equal {Temperature}
variable P equal {Pressure}
velocity all create T 56456546 mom yes rot yes dist gaussian
timestep 1
fix shake all shake 0.0001 20 10000 b 1 3 4
fix 1 all npt temp {T} {T} (100.0dt) iso {P} {P} (1000.0dt)
thermo 1000
thermo_style custom step temp press vol density evdwl ecoul
fix npt_data all print 1000 "{startstep} {t} {pres} d V {evdwl} {ecoul} " file npt.txt
I can see you’ve got SHAKE bonds in your LAMMPS input – are the same bonds held at constant length in GOMC? Also, if you have done a long NVT equilibration run, why would you reset the velocities after that with a velocity command?
Your MD results are likely determined by how you construct your initial geometry.
How have you determined, that the MD systems are properly equilibrated?
Have you compared results from differently constructed (ordered) initial geometries?
Have you compared to results where you are using the final geometry from the MC run as initial geometry of an MD run?
Polymer systems are notoriously difficult to equilibrate with plain MD only (jamming!), so people usually use a mix of MD and MC moves for equilibration. Also, it is not likely making a big difference, but using fix npt with “iso” should only be used for liquids; there is no reason to enforce an isotropic box change on a polymer.
My system is composed of ethane type molecules and not a polymer. So, I think using isotropic changes would be fine for that.
Also, I am not linking the MC & MD runs ; meaning that I don’t use final configuration of MC as initial configuration of MD or vice versa. I am separately running NPT simulations in MC and MD and trying to compare the two densities. That’s where I am getting this undesired difference.
I will try your advice of seeing the difference in result, using different initial geometries. But, I try to start with a box size that is closest to my final expected density at that state point.
I determine the adequacy of equilibration in NVT by seeing the energy vs time behavior of the system over 2 ns.
Finally, do you think in Single Point Energy calculation (using exactly same starting configuration of molecules), 13% pressure difference between MC & MD is something that may have caused this problem?
I dont know the reason why the “single point energy calculations” dont yield the same pressure if the microstate (it’s kinetic energy included) is the same. I am curious to know as well!
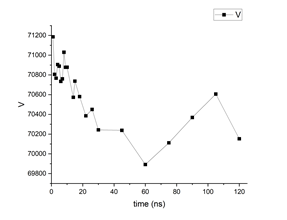
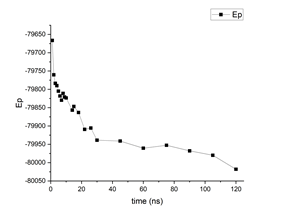
But on what concerns the polymer equilibration, I can show you an example of a polymer (PVDF) simulation that I once did in which I can spot fluctuations in density in the same order of magnitude you mentioned in your initial post. I did in total 120ns of simulation in the NPT ensemble, with the same timestep value as you. Below you can find two graphs showing the evolution of potential energy and volume values. In the plot, the values assigned to a given “time instant” are not actually instantaneous values of the properties: they are the average value of the property within an interval delta_t = 1ns before that the time instant. So for example, the volume value at 1ns in the plot is the average volume during 0ns-1ns.
So you can kind of see that it didnt exactly equilibrate, at least in my case, after 12ns, so maybe, not dismissing the importance of udnerstanding the pressure issue, it would be worth running for a little longer to see what happens next if reproducing the density is such a critical point in your case. Indeed my values in the plot are not averaged over 10ns and I dont know what the effect of initializing boxes of different sizes would have, but at least in my case, depending on the time range, it is still possible to spot fluctuations in the order of what you mentioned if I proceed to calculate the corresponding densities.
My system equilibrated well at the end of 2 ns NVT. It is not a polymer system (its a simple ethane molecule system). Also, my density and energy at the end of NPT simulation (10 ns) was well equilibrated. But yes, I will try to run system for another 10 ns to see if it produces any better results.
I got tricked by the “1,1,1,2 Polytetraflouroethylene” part. So probably running more is indeed useless if you saw that the mean value is indeed steady after all these 12 ns…
When using fix shake, you cannot trust the pressure on step 0. It can be quite far off (for technical reasons that I have explained elsewhere multiple times).
But how can we tell, that your MC results are correct and LAMMPS is incorrect?
How can we know that the settings you are using are really equivalent?
How can we know that you have applied atom typing and assignment of partial charges correctly in both cases?
What is the density that is consistent with the force field parameterization?
I am using mbuild (python pkg) to develop my initial configuration and system for both LAMMPS and MC. Also, I am using the same .xml (force field file) for both. Therefore, assignment of charges, atom typing, forcefield parameters are same for both.
I am using same settings for both software (LJ, cutoff, electrostatics, bond, angle, dihedral styles, reciprocal space, mixing rule, LRC, rigid bonds, temp, pressure values). Only differences are in MD specific constants like timestep, damping constants etc. But that is why I have raised the question here. Maybe there is something that I am skipping somehow which is causing this difference.
Original paper that has details of parameterized forcefield didn’t have NPT density calculations. They did VLE phase diagram. I duplicated that in MC to ensure correction of my parameters/settings which was up to the mark. Then, performed NPT simulations on MC. That is why I am saying that MC is correct and LAMMPS is probably overpredicting.
How large is your system? Try running MC and MD for a 1/8th system (so it’s nice and fast) to see how the discrepancy scales with system length and volume.
Do block standard error analysis for your MC and MD pressure trajectories (Quantifying uncertainty and sampling quality in biomolecular simulations - PMC, 4.8 Statistics). “It looks converged” is always good enough for the person running the simulation and not good enough for Reviewer #2 – a BSE graph is good quantitative evidence for convergence (and more than most researchers do, frankly, which is shocking given how little it costs).
Try the mtk yes keyword in fix npt.
For your single point pressure calculation, try using a short nve run with very small timestep (say dt = 0.1) to shake off the SHAKE first step problem, then compare the pressure at the final configuration with GOMC’s single point calculation.
And so on. The general idea is that right now we know as much as you do about why LAMMPS is giving a different value from GOMC (that is, not very much), so you need to do more tests to develop an actionable hypothesis.
One more thing to check is the LJ smoothing function. Lennard-Jones with a simple cutoff (lj/cut) has a force discontinuity at the cutoff, and many force fields try to introduce a smoothing function between this “outer” cutoff and an “inner” cutoff so that the force smoothly goes to zero in between those regions. You did not explicitly mention this in the list of things you checked – if you have checked this, then it wouldn’t be an issue.
I am obliged for all your valuable input. In my MC and original paper, it is LJ cut with no smoothing. Therefore, I did not introduce that in Lammps.
I am using 1000 molecules for both MC & LAMMPS. I used this size to rule out finite size effects. But, sure I can run smaller systems to see the behavior.
Do you recommend doing BSE for equilibration part or final production run?
I did step 0 pressure comparison without shake. Before it was 13%. Without shake, results are way off like 93%. I don’t know what to make of it.
PS: In the meantime, I also ran same simulation on GROMACS with same build input file and all the same parameters. And I got exact density as MC. Difference is 0.1%.
I am pretty sure I am doing something wrong in LAMMPS.
Which LAMMPS version are you using? Are you using any acceleration packages (INTEL, GPU, Kokkos, etc)? Did you compile LAMMPS yourself or are you using something compiled by admin on a cluster?
Can you post a data file with just one molecule? In particular, it’s a bit curious that you have 4 bond types – I would have expected only three for R134 (C-H, -F and -C).
Do you have any dangerous neighbor builds? I can see your settings of “neighbor 6.0” in your input script – can you set that back to the default of 2.0?
Since we want to know about convergence for both parts, you should try it for both parts.
Well yes the pressure with SHAKE will be very different to the pressure without since rigidifying the bonds completely changed the system’s equations of motion.
Another thought about the “single point pressure calculation”: LAMMPS calculates the temperature contribution via a temperature compute (see documentation for compute pressure), which is directly calculated from the system’s velocities. But an MC simulation has no velocities and so that component of pressure would just reuse the input temperature. If you calculate temperature and pressure simultaneously in LAMMPS you can adjust for the instantaneous difference between preset and actual temperature and see if that makes a difference. (For that matter, does your thermo output temperature match what you imposed in fix nvt)?
AND have you got the right pressure units?? GOMC and GROMACS take pressure in bars, but LAMMPS units real specifies pressure in atm. 1 atm = 1.013 bar, which would correspond to 1.3% more pressure.
But that’s not the LAMMPS data file, which should specify the bond coefficients for each type and the bond topology (which bonds connect which atoms, and what type each bond is). The mol2 file you attached does indeed have only three bond types, not four, so it’s still not clear what is happening when you supply b 1 3 4 to fix shake.