Hello everyone,
I’m working on annealing simulations of fluorinated diamane (F-diamane) using LAMMPS and ReaxFF. I’ve run into some puzzling differences between a smaller and a larger replicated system, and I thought I’d seek feedback on the potential reasons behind this behavior, as I’ve viewed posts here that discuss how stable/“constant” temperature and pressure are attributed to big systems.
My work is as follows: an initial structure of an F-diamane slab with fluorine fully terminating both sides. A small system I created consisted of 384 atoms (256 C + 128 F), and I created a larger system repeating the smaller unit cell in the x- and y-directions, consisting of 3456 atoms (2304 C + 1152 F). Below is the script I use for annealing:
# 1) INITIALIZATION
# ------------------------------
variable T_start equal 10.0 # Starting temperature
variable T_end equal 773.15 # Annealing target temperature
units real
atom_style charge
boundary p p p
read_data f_diamane_big.data
# ------------------------------
# 2) INTERACTION MODEL
# ------------------------------
pair_style reaxff NULL safezone 3.0 mincap 150
pair_coeff * * ffield.reax.FC.txt C F
# ------------------------------
# 3) GROUPS & CHARGES
# ------------------------------
group carbon type 1
group fluorine type 2
group all_atoms union carbon fluorine
fix qeq all_atoms qeq/reaxff 100 0.0 10.0 1.0e-6 reaxff
# ------------------------------
# 4) PRE-ANNEAL RELAXATION
# ------------------------------
fix boxrelax all_atoms box/relax x 0.0 y 0.0 vmax 0.001
minimize 1.0e-6 1.0e-9 1000 10000
unfix boxrelax
velocity all_atoms create ${T_start} 4928459 mom yes rot yes dist gaussian
# ------------------------------
# 5) PHASE 1: Temperature Ramp
# ------------------------------
fix temp_ramp all_atoms nvt temp ${T_start} ${T_end} 50
timestep 0.25
thermo 100
run 10000 # 2.5 ps temperature ramp
unfix temp_ramp
# ------------------------------
# 6) PHASE 2: Hold at T_end
# ------------------------------
fix anneal all_atoms nvt temp ${T_end} ${T_end} 50
fix reax_bonds all_atoms reaxff/bonds 100 bonds.reax
dump traj all_atoms custom 500 dump.lammpstrj id type x y z q
run 90000 # 22.5 ps hold at 773.15 K
# Total simulation time = 25 ps
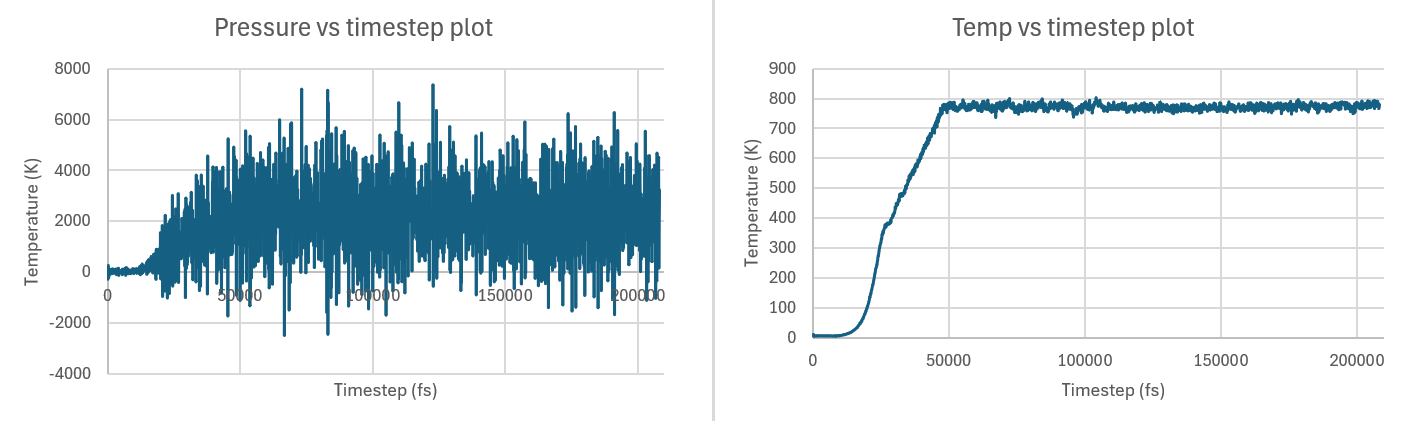
Below are the respective temperature and pressure plots for the simulation for each system:
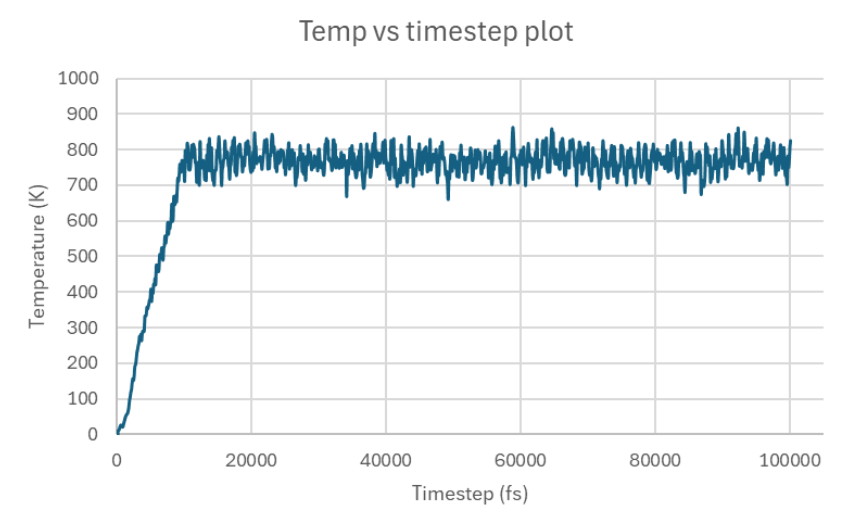
Small system clearly showing non-convergence, and so I thought of running the same tests on a bigger system:
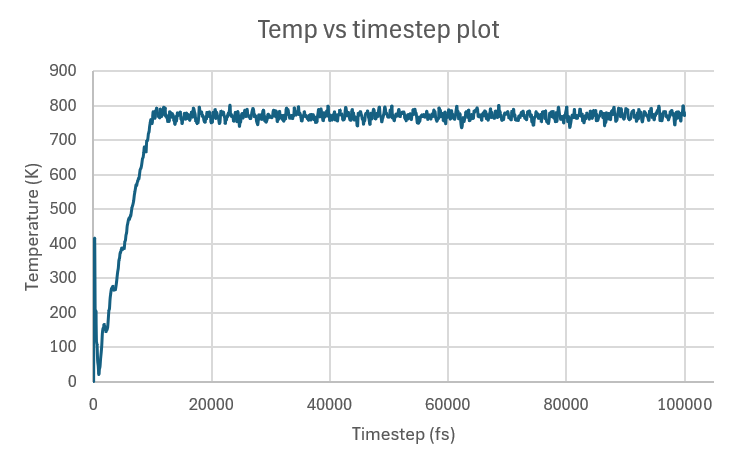

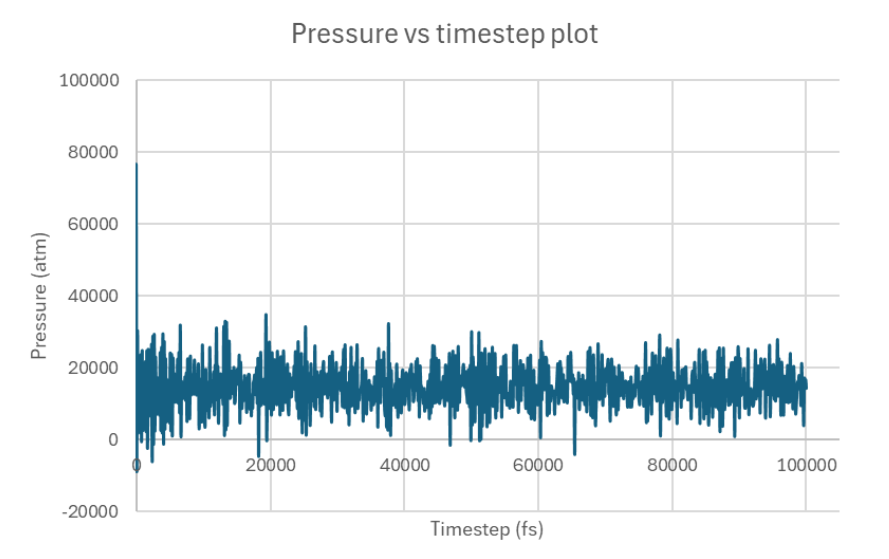
Note that the pressure plot was truncated to better display the behavior at the beginning of the run. The rest of the run exhibited smooth trends like the ones shown after the initial fluctuations.
I’m happy for the most part with the temperature trends of the big system, as temperatures are more tightly packed or don’t fluctuate as much. The one concern I have is the initial temperature spike, which I believe is attributed to really high pressure:
Step Temp E_pair E_mol TotEng Press
22 10 -502971.03 0 -502868.04 -12120.994
--> 100 365.77929 -511393.37 0 -507626.32 48195.76
200 413.21375 -514338.02 0 -510082.46 -6067.4625
300 160.32654 -513229.38 0 -511578.22 -14845.023
400 204.45019 -514380.16 0 -512274.59 2087.0516
500 115.7196 -514030.76 0 -512839 6686.7493
600 106.61311 -514253.49 0 -513155.51 12875.709
700 60.446976 -513999.85 0 -513377.32 -34039.995
800 45.34627 -513956.07 0 -513489.07 19513.915
900 19.933844 -513774.81 0 -513569.52 -11997.832
1000 35.513314 -513879.51 0 -513513.77 20665.207
1100 43.236585 -513853.29 0 -513408.01 -41481.65
1200 66.191752 -513891.15 0 -513209.46 18514.136
1300 83.267452 -513866.3 0 -513008.76 -7292.2423
1400 105.54025 -513858.32 0 -512771.39 7358.7768
despite attempting to avoid this issue which I encountered with different simulations in the past by using fix box/relax to rescale the simulation box and resolve any potential in-plane strain.
Onto pressure trends, they seemed to inevitably settle down/converge but I’m curious about the cause of the initial fluctuations and if they’re attributed to my “big” system not being that big, bad practice/scripting on my part, or an alternate reason.
— LAMMPS version: 29 Aug 2024 - Update 1, on Windows
Thank you in advance and apologies for any oversights or misconceptions