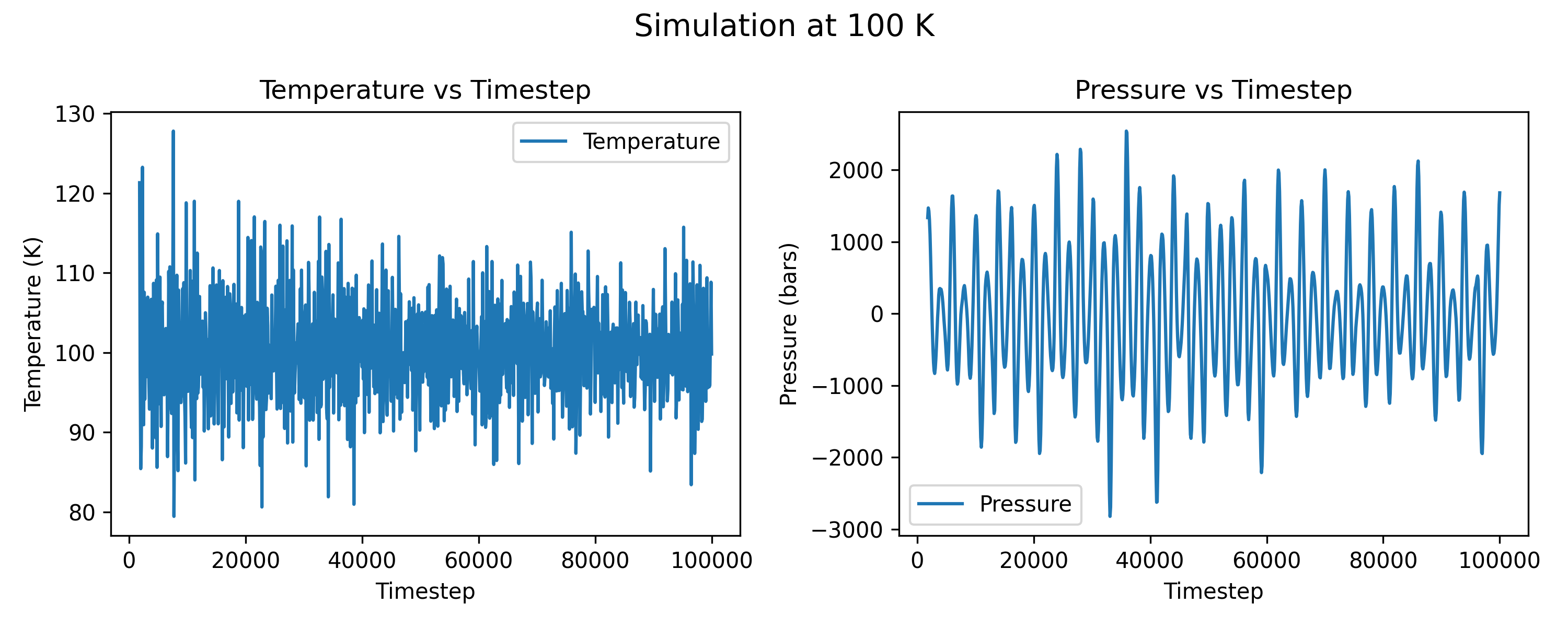
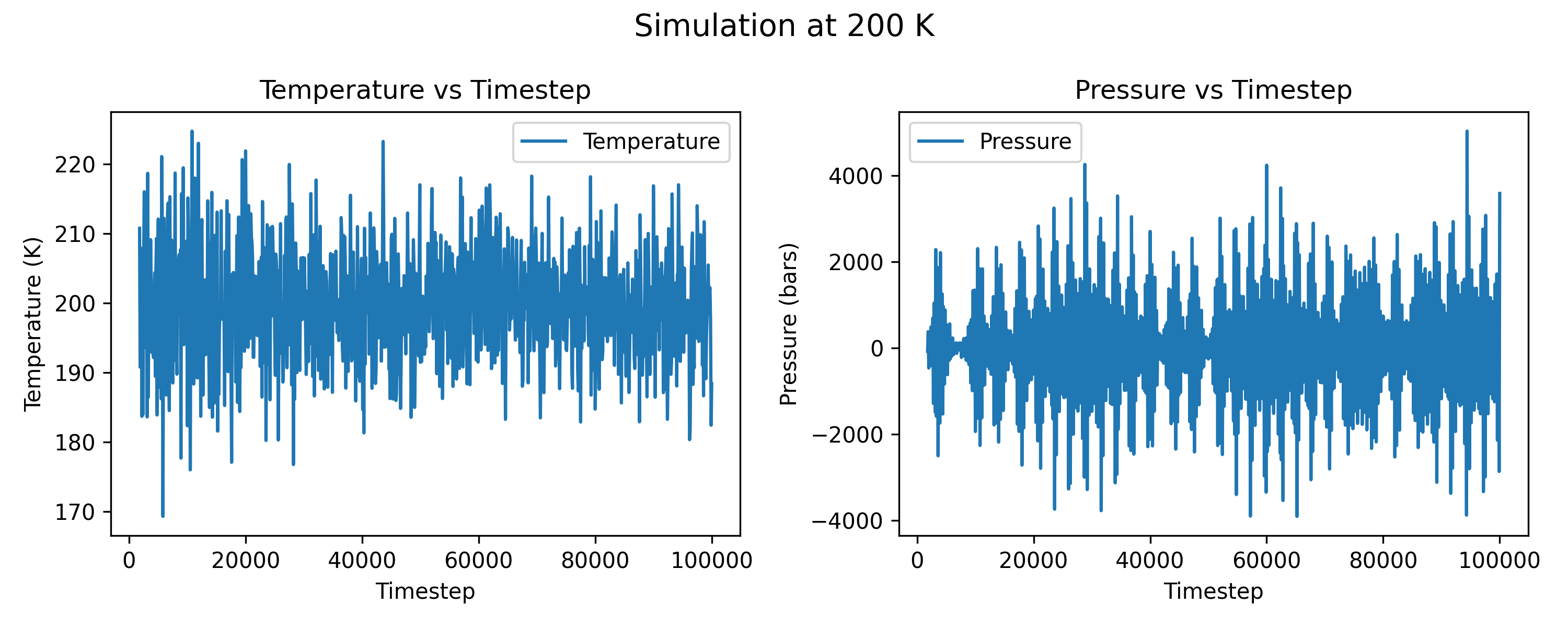
I am running simulations with LAMMPS with npt for germanium in a diamond structure. This bulk is a 4 * 4 * 4 supercell, containing 512 atoms, and the potential is MEAM. The main commands that I use for the attached plots are:
units metal
timestep 0.0005
fix npt all npt temp ${T} ${T} $(100.0*dt) iso 0.0 0.0 $(500*dt)
run 100000
${T} is the temperature, here in my plots, I use 100 and 200 K.
I tried different Pdamp parameter with other values (1000 * dt, 5000 * dt), but the pressure seems cannot converge. For all these Pdamp settings, the absolute fluctuation value is over 1000 bars. Besides, I think the simulation bulk is large enough, and the timestep is reasonable. I’m wondering if there’s any suggestions? Thanks in advance.
What you “think” is irrelevant. This is a topic covered by statistical mechanics and thermodynamics, so whatever laws and rules have been devised in that context apply.
A system of 512 atoms is tiny and a “constant” temperature or pressure only applies in the limit of an infinite number of atoms. There have been many previous discussions on this subject, so you can read and learn from those.
Thanks for the reply! Actually I checked the previous discussions before I created the new topic. There are mainly three factors related to the pressure convergence: compressibility, system size, and choice of time step. I’m a bit confused, based on the current results, which parameter I should change. I’ll think and test more carefully. Thanks anyway!
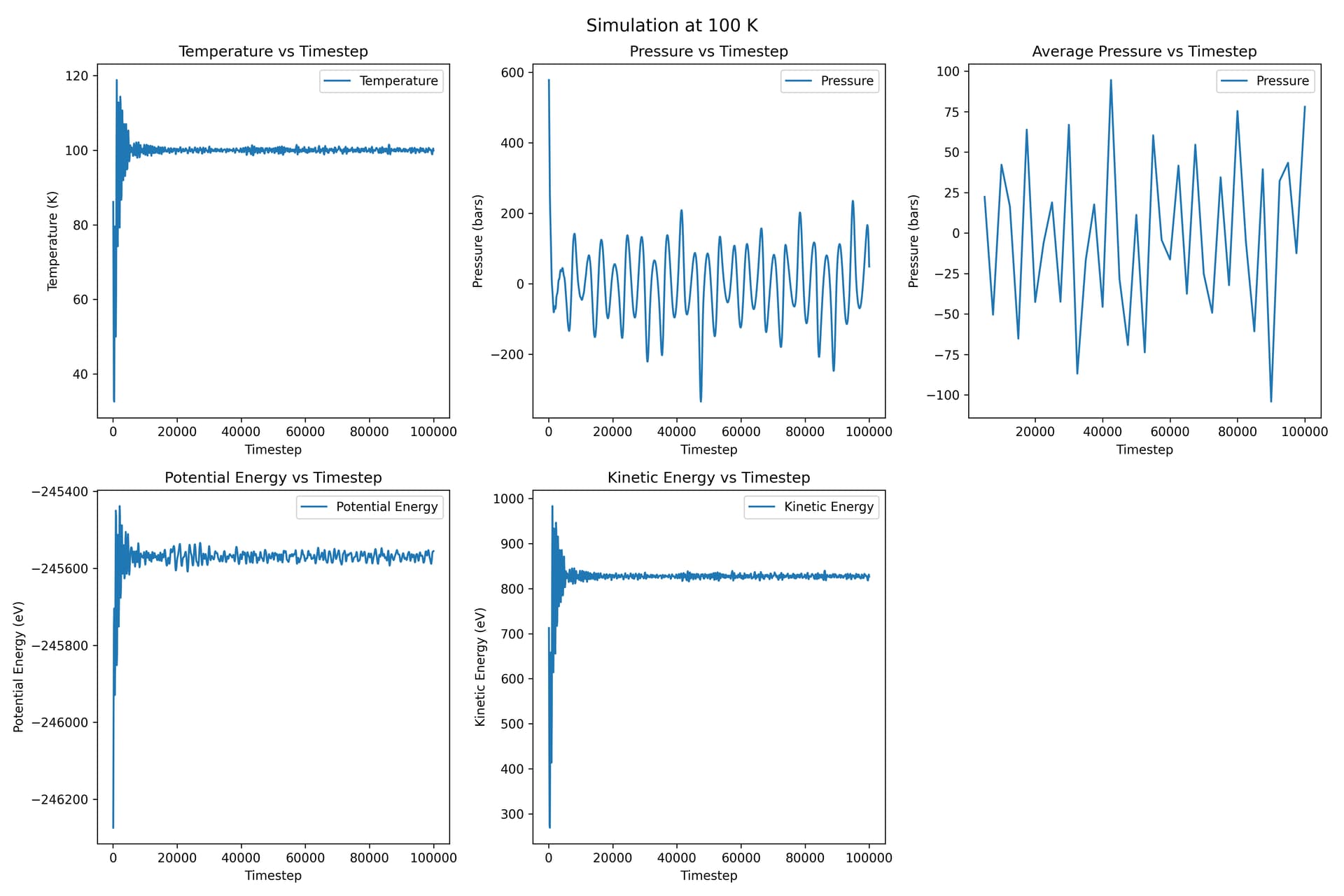
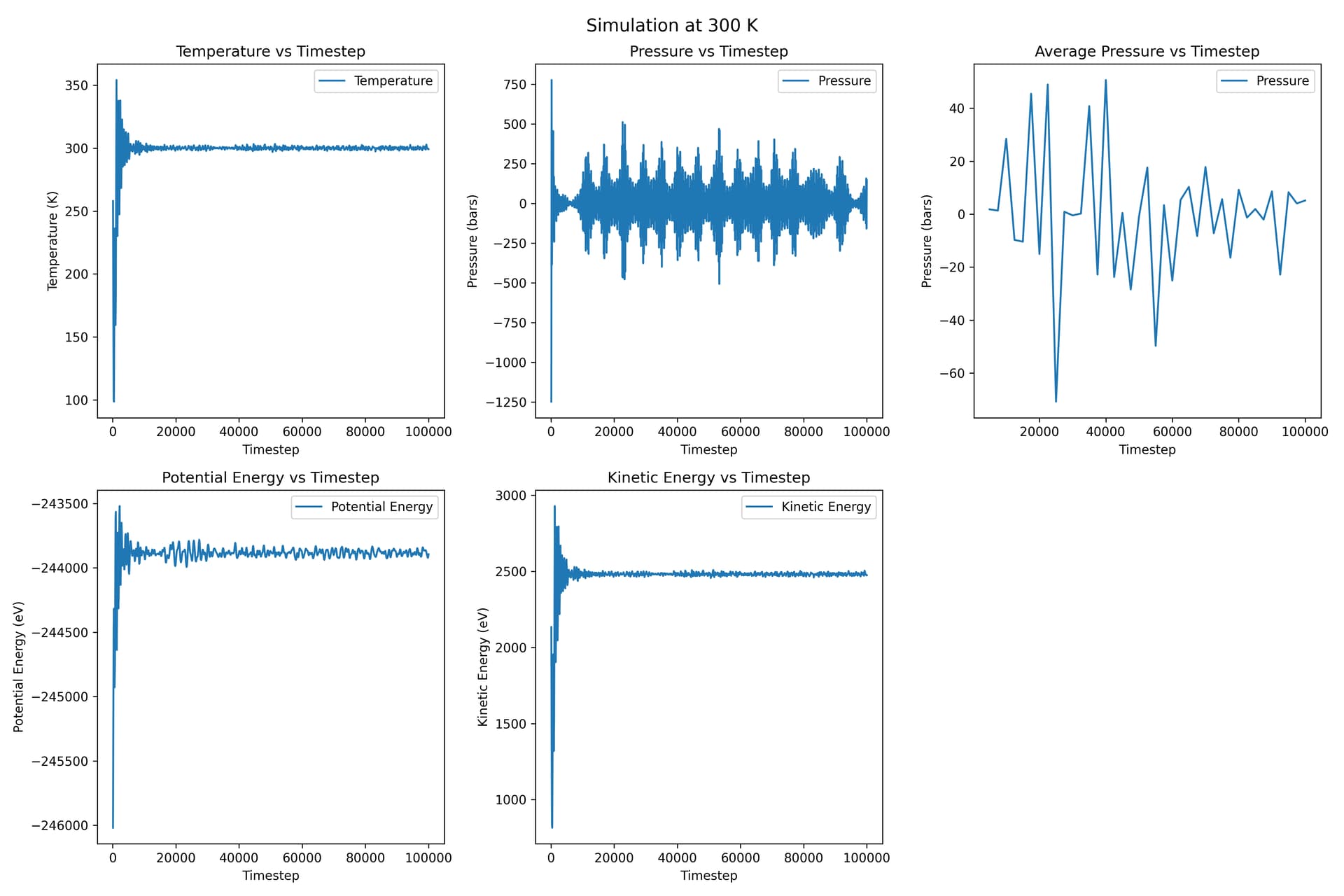
Thanks for your reply! I tried this method, and my command is fix ave_output all ave/time 500 5 2500 v_mypress file Results-MEAM/${T}/ave_press.txt
Besides, this time I used a 20 * 20 * 20 supercell containing 64000 atoms.
my main commands are:
units metal
timestep 0.0001
fix npt all npt temp ${T} ${T} $(100.0*dt) iso 0.0 0.0 $(1000.0*dt)
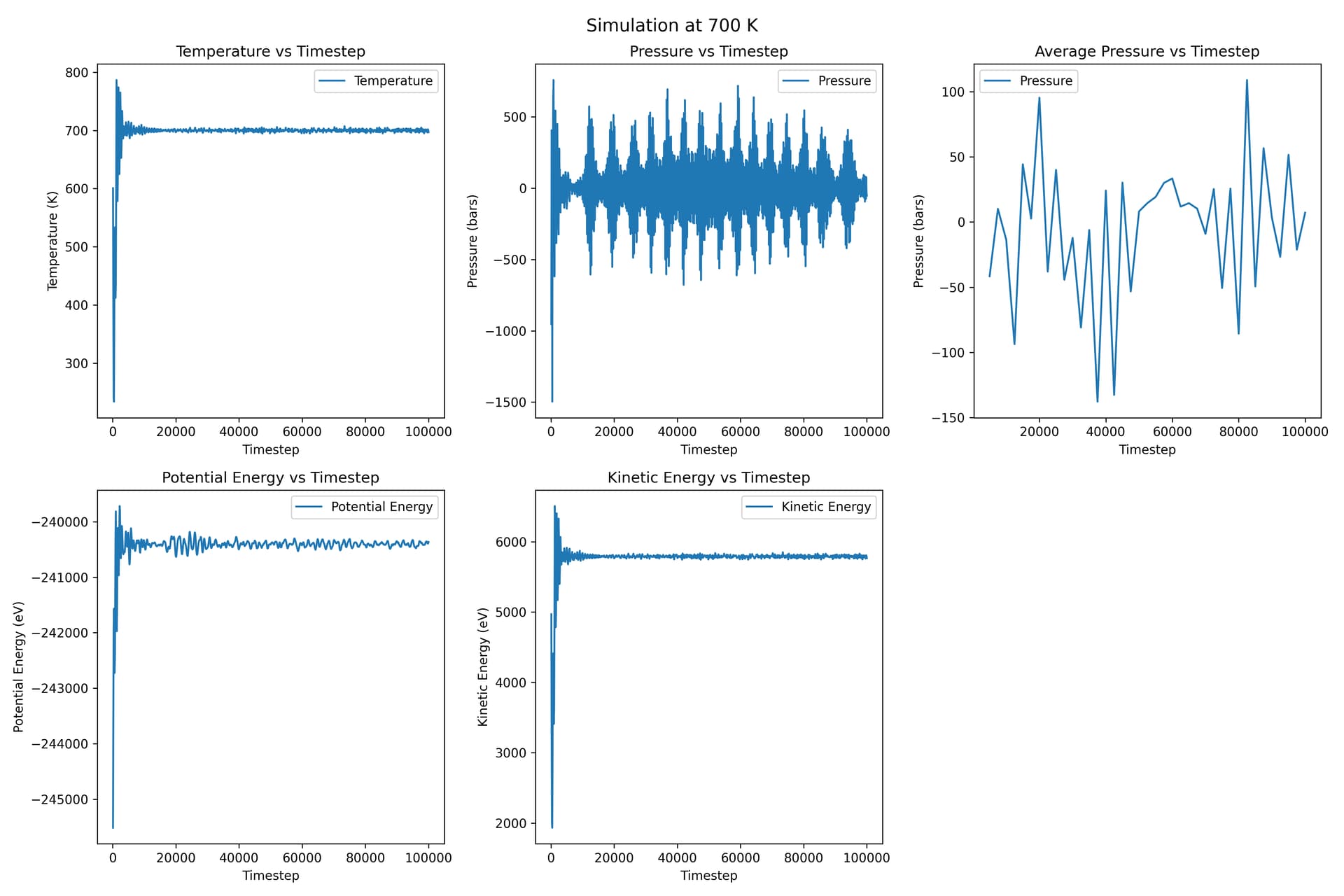
For each temperature plot, the ‘average pressure vs timestep’ is the plot for average pressure, and the one on its left is the original pressure vs timestep.
This time the pressure fluctuations are much smaller.
All thermodynamic properties like T and P are averages. None can be defined instantaneously for an atomic system. It’s helpful to remember that the things you are plotting are not the thermodynamic T and P, but their averages are.
May I ask a further question? I updated my results above, where I replied to another Professor. This time I used a 20 * 20 * 20 supercell, and the magnitude of fluctuations becomes smaller. I’m not very sure, if I want to calculate elastic constants under these temperatures, I need to run NPT simulation at these temperatures with a large supercell so that the system can get equilibrium, and then I calculate elastic constants at the corresponding temperature, right? Thanks in advance.
One further problem you have is that if you are simulating a regular crystalline structure, you should expect to frequently see regular harmonic oscillations as a consequence of thermal vibration of phonons. I don’t know what the answer to this is, but it is very unlikely to involve barostatting, especially with a Nose-Hoover barostat since the Nose-Hoover approach is known to struggle with making harmonic dynamics ergodic.