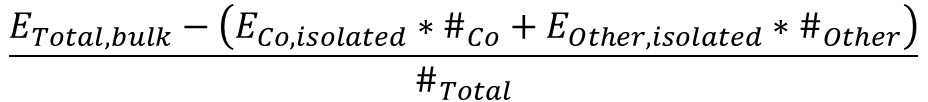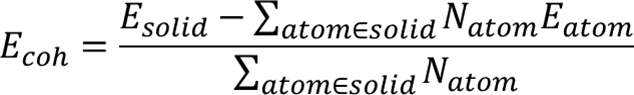Hello.
I’m trying to extract cohesive energy from the materials project database by using API.
I tried reading answers to similar questions and following those methods, but unfortunately, I couldn’t succeed. It’s because the pymatgen was updated to the latest version, and the get_cohesive_energy function that used to be in the old version has disappeared.
Currently, with the materials project being renewed, it seems that using the existing pymatgen.ext.matproj for get_cohesive_energy isn’t possible. I’ve actually tried several times, and I keep running into the following error:
cohesive = mpr.get_cohesive_energy(self, ‘mp-1206874’, per_atom=True)
^^^^^^^^^^^^^^^^^^^^^^^
File “/home/{my_folder_name}/anaconda3/lib/python3.11/site-packages/mp_api/client/mprester.py”, line 399, in getattr
raise AttributeError(
AttributeError: ‘MPRester’ object has no attribute ‘get_cohesive_energy’
It seems that the newest version of pymatgen (24.10.3) no longer have the get_cohesive_~ part.
Is there any other way to retrieve the cohesive energy? I think there’s also a method using entry (pulling total energy and dividing by the number of atoms), but this wouldn’t give an accurate calculation for isolated atoms, making the reliability of the results questionable.
I’ve finished calculating the isolated energy for each atom, but there’s a big problem. There’s a huge difference between the total energy from the Materials Project data calculated with VASP and my data calculated with Quantum ESPRESSO. I found out that the total energy can change depending on the calculation method, so it doesn’t have physical significance, but I still don’t understand how this difference occurs. Also, if I can’t directly calculate the difference in total energy, I have no idea how to compare the calculated values between VASP and QE.
Please let me know if there’s a good method available. I really debated whether to call back this topic for several days and tried various things, but it’s just not working out as I hoped…
One sure way to handle this would be to downgrade pymatgen version which contain that codes, but unfortunately, due to some errors, downgrading is currently not an option.
[Addtional]
Can anyone explain the options for predicted stable and Energy_above_hull? From what I understand, to be predicted stable, the energy above hull should be 0. I’ve checked materials like Co, Ru, and Cu, and it turns out that only those with an energy above hull of 0 are considered predicted stable. I’m curious if there’s a specific reason for separating these two options.
Best regards.
Code that I used.
import os
from mp_api.client import MPRester as MP_API_MPRester
from pymatgen.ext.matproj import MPRester as Pymatgen_MPRester
API_KEY = “”
with MP_API_MPRester(API_KEY) as mpr:
structures = mpr.materials.summary.search(
elements=[“Co”],
chemsys=“Co-*”,
energy_above_hull=(0, 0),
is_metal=True,
is_stable=True
)
with open(“2cohesive_test.txt”, “w”) as file:
file.write(f"Number of documents: {len(structures)}\n\n")
if not structures:
file.write("No results found.\n")
else:
with Pymatgen_MPRester(API_KEY) as pmg_mpr:
for idx, structure_summary in enumerate(structures):
material_id = structure_summary.material_id
formula = structure_summary.formula_pretty
sym = structure_summary.symmetry
structure = pmg_mpr.get_structure_by_material_id(material_id)
primitive_cell = structure.get_primitive_structure()
cif_filename = f"{formula}.cif"
primitive_cell.to(filename=cif_filename)
try:
bulk_entry = pmg_mpr.get_entry_by_material_id(material_id, conventional_unit_cell=True)
if isinstance(bulk_entry, list):
bulk_entry = min(bulk_entry, key=lambda e: e.energy_per_atom)
bulk_energy = bulk_entry.energy
isolated_atom_energies = {}
for element in bulk_entry.composition.elements:
if element.symbol == 'O':
o2_entries = pmg_mpr.get_entries('O2')
if not o2_entries:
raise ValueError("Unable to find O2 molecule entry.")
o2_entry = min(o2_entries, key=lambda e: e.energy_per_atom)
isolated_atom_energy = o2_entry.energy_per_atom
isolated_atom_energies[element.symbol] = isolated_atom_energy
elif element.symbol == 'H':
h2_entries = pmg_mpr.get_entries('H2')
if not h2_entries:
raise ValueError("Unable to find H2 molecule entry.")
h2_entry = min(h2_entries, key=lambda e: e.energy_per_atom)
isolated_atom_energy = h2_entry.energy_per_atom
isolated_atom_energies[element.symbol] = isolated_atom_energy
else:
element_entries = pmg_mpr.get_entries_in_chemsys([element.symbol])
bulk_element_entries = [
e for e in element_entries
if e.composition.reduced_formula == element.symbol
]
if not bulk_element_entries:
raise ValueError(f"Unable to find bulk element entry for element {element.symbol}")
bulk_element_entry = min(bulk_element_entries, key=lambda e: e.energy_per_atom)
isolated_atom_energies[element.symbol] = bulk_element_entry.energy_per_atom
total_isolated_energy = sum([
bulk_entry.composition[el] * isolated_atom_energies[el.symbol]
for el in bulk_entry.composition.elements
])
cohesive_energy = total_isolated_energy - bulk_energy
cohesive_energy_per_atom = cohesive_energy / bulk_entry.composition.num_atoms
except Exception as e:
cohesive_energy_per_atom = None
print(f"Can't calculated {material_id} ({formula})'s cohesive energy: {e}")
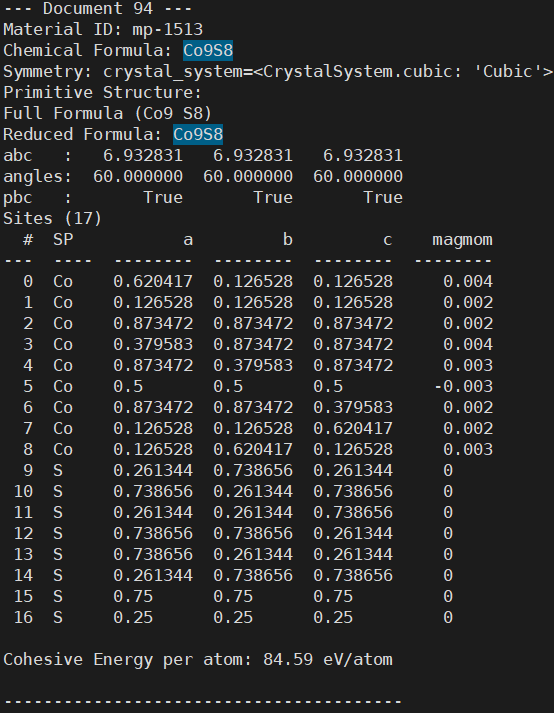
file.write(f"--- Document {idx + 1} ---\n")
file.write(f"Material ID: {material_id}\n")
file.write(f"Chemical Formula: {formula}\n")
file.write(f"Symmetry: {sym}\n")
file.write(f"Primitive Structure:\n{primitive_cell}\n")
file.write("\n")
if cohesive_energy_per_atom is not None:
file.write(f"Cohesive Energy per atom: {cohesive_energy_per_atom:.2f} eV/atom\n\n")
file.write(f"Bulk Energy: {bulk_energy:.4f} eV\n")
file.write("Isolated Atom Energies (DFT calculations):\n")
for element_symbol, energy in isolated_atom_energies.items():
file.write(f" - {element_symbol}: {energy:.4f} eV per atom\n")
file.write("\n")
else:
file.write("Can't calculate cohesive energy.\n\n")
file.write('-' * 40 + "\n")
file.write("\n\n")