I don’t simply want an initial starting structure, what I did is just a
test. But insertion is an important point of my research, I would be
grateful if someone would solve this issue.
My apologies.
What you are trying to do is difficult.
The “minimize” command, in its current form, is not ideal for what you are trying to do (unless you try modifying the forces in your system).
In my experience, the “minimize” command usually fails when there are extremely large forces in your system (for example when using 1/r^12 Lennard-Jones-style repulsion). The “minimize” command will fail unless the distance between all pairs of atoms are at least 10%(??) of the sum of their their radii. (It’s not possible to estimate the exact distance when failure occurs, but it definitely happens at non-zero distances.) This means if you insert a molecule randomly with respect to the other molecules, eventually you will crash LAMMPS. Even if the system is dilute, eventually you will have a collision that causes a pair of atoms to be too close together. I am not sure whether this can be alleviated by modifying the source code for the “minimize” command. I suspect it is an inherent limitation of double precision floating point and 1/r^12.
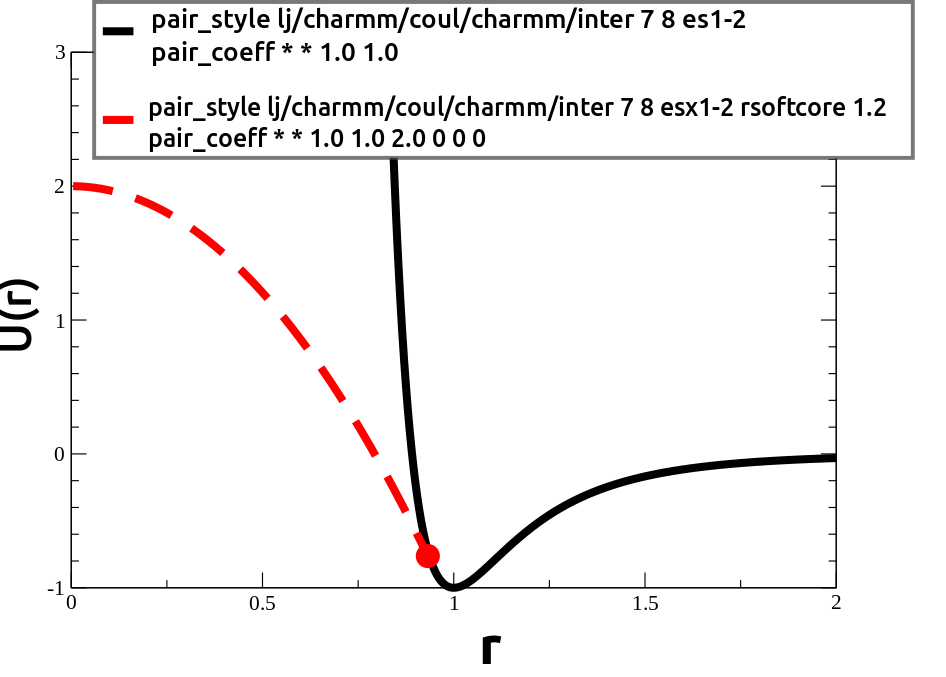
To help get around these problems, for the moment I have been using pair_styles which do not necessarily diverge to ∞ as r → 0. (Such as pair_style soft, pair_style gauss, or pair_style lj/charmm/coul/charmm/inter, which you can currently download here. See the example using “rsoftcore” here. Let me know if that code no longer compiles. It’s kindof funky.)
Using soft potentials does not always solve the problem, but it seems to help.
In this case, it should be either possible to minimize the system using the “minimize” command, or run an ordinary MD simulation with a small timestep and high damping (using some combination of “fix langevin” and “fix nve”). If you attempt molecule insertion every 10000 timesteps, then you will have to write a loop which minimizes the system, right after the insertion every 10000 timesteps (perhaps changing the pair style each time).
---- other issues that make minimization difficult (after insertion) ----
- The “minimize” command effects all of the atoms in your system. (There is no “group” argument) The entire simulation has to halt and wait for minimization to finish. In your case, it could be more efficient to only minimize atoms which are located nearby where you inserted the molecule. Doing this would require writing some kind of new fix, or (better) some function which other fixes can invoke after a sudden change in the system(such as fix bond/create, fix bond/break). I’m not yet certain whether it would be worth the effort. It depends on how frequently these insertions occur, and in how many different locations.
I run a lot of simulations here with bonds being created and destroyed. I have to use small timesteps to avoid numeric explosions each time I add or delete a bond. At some point, I may decide to attempt writing a minimizer which solves some of these issues, but I haven’t had the time to focus on the issue yet. I recently saw an interesting discussion regarding the new “fix bond/react” feature being contributed by Jacob Gissinger. This fix a group of atoms which are effected every time a bond is created or destroyed. (But I forget the details.) Perhaps he eventually planned to implement a minimizer that would act on only this group? In the distant future, I was considering creating a new fix based on my code or his code which would create or insert molecules at locations depending on the nearby environment. Perhaps we should collaborate.
- The “minimize” command is not compatible with constraints like “fix rigid”. (Perhaps this is not an issue for you.)
- Both minimization procedures in case 1 and case 2 give the similar values
of those energies. The program crashes during the dynamic procedure.
In case 2, the minimization after insertion gives E_pair = -12414.855 at
10707 step, but in the dynamic, the E_pair suddenly changes to 1450.7209 at
10707 step. In case 1, E_pair doesn’t change after minimization.
I’m not sure why. Again, weird behavior like what you are reporting is common in LAMMPS when using the “minimize” command with overlapping particles.
It used to bother me, but now I have gotten used to it.
I would guess that E_pair = -12414.855 is true at the beginning of the timestep (Epair(x(t)) and E_pair=1450.7209 at the end of that timestep (Epair(x(t+Δt)).
Displaying the energy (printing thermo to terminal or log.lammps) is not done until after the timestep has completed, which includes code which causes the particle to move. (See page 14 of http://lammps.sandia.gov/doc/Developer.pdf)
To test whether this is the reason, try setting the velocity of all of the atoms to 0 before using the “run” command", and try using the simplest possible integrator such as “fix nve” (not “fix nvt” or “fix npt”). Also try reducing the timestep to 0 (or a very small value if LAMMPS won’t allow timestep=0).
{kind=link}