Hello, I am seeking your help to share the EAM potential file for the WC-Co, W-W, W-C, W-Co, C-C, C-Co, and Co-Co models. Thank you very much for considering my request and for your willingness to assist.
I don’t think that any of the people that regularly respond here spend time on parameterizing potentials.
Thus the first place to look for potentials is the published literature.
My gut feeling is, however, that your compound has enough directional interactions that EAM is not sufficient to describe the interactions well. So, more likely, you will have to look for a set of MEAM potential parameters or something else like some machine learning potential. That would also mean that your chances are slim to find such a potential, since it is difficult and/or laborious to do. So you may be left with either finding a different subject of study or having to learn how to properly parameterize potentials first.
Thank you so much for your suggestion.
Hello again,
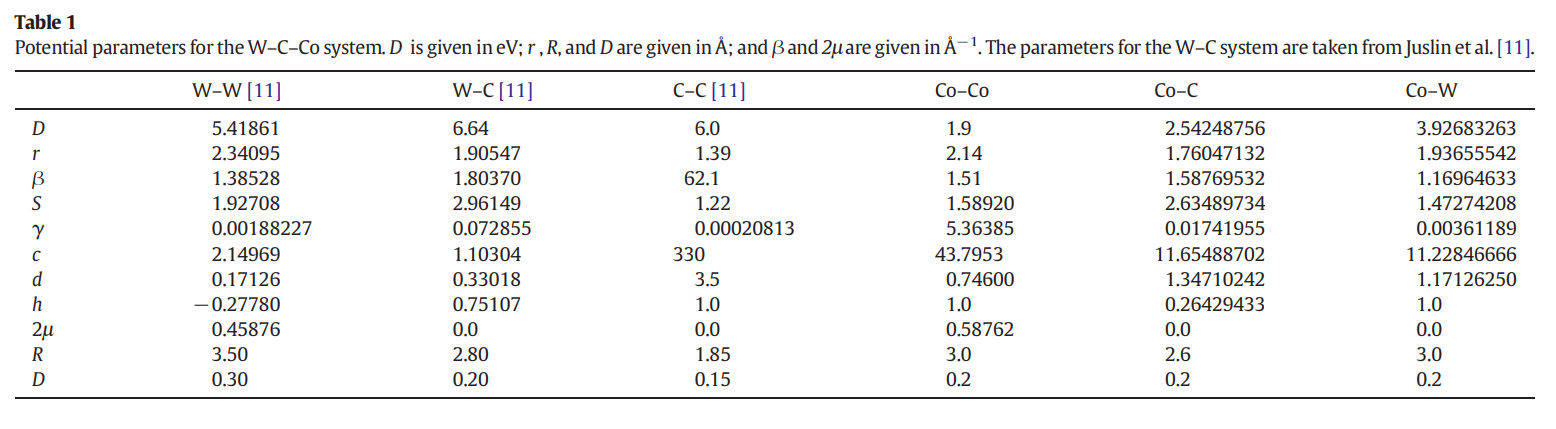
Following your suggestion, I referred to similar study papers (Redirecting) and managed to obtain the potential parameters for WW, WC, WCo, CC, CCo, and CoCo, as shown below. These parameters appear to closely align with ABOP parameters.
My question is: Do I need to convert these ABOP parameters to ABOP potential and then to Tersoff potential, or how should I use them for my LAMMPS analysis?
Thank you very much for considering my request and for your willingness to assist.
There has been a similar discussion here.
Hello Dear Representative and Members,
Based on the parameters above, I have formulated the WCCo.tersoff file. I validated the elastic properties, and they match the results found in the referenced paper. Therefore, I am proceeding with this Tersoff file.
However, I am encountering some difficulties in creating the interface between FCC-Co and Bh-WC.
This is the structure before relaxation:

And this is the structure after relaxation:

I have tried to optimize the simulation box size to achieve the minimum lattice constant, but there still appears to be some stress. I am concerned about proceeding with further analyses using this structure.
After many trials, I achieved a somewhat stable box dimension from (0 0 0) to (24.575118 68.915334 104.2634). However, some atoms at the interface between WC and Co move their positions after relaxation. I used the atom positions of WC and Co as follows:
region whole block 0.000000 {xlength} 0.000000 {ylength} 0.0 ${zlength} units box
create_box 3 whole
region latlower block INF INF INF INF 0.000000 ${zhalf} units box
variable b equal (1/2)*sqrt(3)
variable c equal 2/3
variable d equal 1/3
variable e equal 2.81157226240964/2.91693096603313
lattice custom 2.91693096603313 &
a1 0.5 -${b} 0 &
a2 0.5 ${b} 0 &
a3 0 0 ${e} &
basis 0.0 0.0 0.0 &
basis ${c} ${d} 0.5
create_atoms 1 region latlower basis 1 1 basis 2 2
group latlower region latlower
region latupper block INF INF INF INF {zhalf} {zlength} units box
lattice fcc 3.53848314880901 orient x 1 -1 0 orient y 1 1 -2 orient z 1 1 1 spacing {dx} {dy} ${dz}
create_atoms 3 region latupper
group latupper region latupper
I would appreciate any suggestions on whether I should proceed to calculate the interfacial energy and other properties with this structure, or if further improvements in the simulation box size are necessary (if it affect the result).
Thank you.
Fetih
First of all, congratulations! This is the way to proceed. But you can do better by editing your post to properly quote the input script (tip: press the </> button).
The relaxation of atoms at the interface is to be expected, but the magnitude of it is affected by the finite size of the simulation box. The best you can do is to optimise the crystal structures of the two phases separately using an NPT integrator, including the off-diagonal components of the pressure tensor. From the relaxed structures, compute the average crystal cell vectors and use them to find the surface vectors of the planes you want to match. From these vectors, you could write a small program to find a supercell that accommodates both. I would test different choices of the supercell to see how that affects the interfacial energy.
Thank you so much for your reply and suggestion, I did and now the interface is perfect.
Now I created vacancy by considering only the upper part (only pure Cobalt) to see the diffusion coefficient at higher temp (1000K) and to visualize the vacancy moment. However, the vacancy can’t move, and i extract energy barrier 0eV.
your advice on the fix command and overall script appreciated. I used the LAMMPS version (20 Nov 2019) Here i attached the script:
dimension 3
boundary p p p
atom_style atomic
atom_modify map array
# ---------- Create Atoms ---------------------
region box block 0.0 24.57512 0.0 68.915334 -1.0 58.2455005 units box
create_box 1 box
lattice fcc 3.53848314880901 orient x 1 -1 0 orient y 1 1 -2 orient z 1 1 1
region Co block 0.0 24.57512 0.0 68.915334 -1.0 58.2455005 units box
create_atoms 1 region Co units box
# ---------- Define Interatomic Potential ---------------------
pair_style tersoff
pair_coeff * * CCoW.tersoff Co
neighbor 2.0 bin
neigh_modify delay 10 check yes
# ---------- Create a vacancy by deleting one atom in the center ----------
group DEL_ATOM id 4343
delete_atoms group DEL_ATOM compress yes
# ---------- Assign mass to cobalt atoms ----------
mass 1 58.933
# Minimization
fix 1 all box/relax iso 0.0 vmax 0.001
min_style cg
minimize 1.0e-12 1.0e-12 50000 50000
unfix 1
# Initialize velocities
velocity all create 1000 2547321 mom yes rot yes
# Equilibration
fix 2 all npt temp 1000.0 1000 0.01 iso 0.0 0.0 1.0
thermo 1000
run 200000
unfix 2
reset_timestep 0
# Main run with MSD calculation
fix 3 all nve
compute msd all msd com yes average yes
variable twopoint equal c_msd[4]/6/(step*dt+1.0e-6)
fix 9 all vector 100 c_msd[4]
variable fitslope equal slope(f_9)/6/(10*dt)
fix 4 all ave/time 10 10 100 c_msd[1] c_msd[2] c_msd[3] c_msd[4] file average-msd_Co_1_Vacancy_1000K.txt
# Output settings
thermo 10000
thermo_style custom step temp pe etotal pxx pyy pzz lx ly lz c_msd[1] c_msd[2] c_msd[3] c_msd[4] v_twopoint v_fitslope
log Co_1_Vacancy_1000K.txt
# Dump atom positions for visualization
dump 1 all custom 100000 Co_1_Vacancy_1000K.lammpstrj id type x y z
# Main run
run 3000000
unfix 3
And yet again you are not formatting your post correctly. Where is the problem in making an effort so that it is easier (or at all possible) to help you? Just look at your post, can you read all of it properly?
After all, it is you that is looking for help. If you don’t respect the guidelines and wishes of the people responding here, how can you expect that somebody will help you? And what if somebody tries to guess and guesses wrong and thus gives you bad advice?
Sorry for the late reply, Dr. Axle.
In my previous post, I was about to make some edits, but it wasn’t editable. I absolutely accept and respect the guidelines and the representatives. I apologize for not editing the previous post as intended.
I edited the current post by enclosing the text with three backquote characters and mentioned the LAMMPS version.
In my current case, I created a vacancy in FCC Cobalt to study the diffusion coefficient at a higher temperature (1000K) and to visualize the vacancy movement. However, the vacancy does not move, and I am extracting an energy barrier of 0 eV. I am wondering if you could provide some suggestions for improving the script.
dimension 3
boundary p p p
atom_style atomic
atom_modify map array
# ---------- Create Atoms ---------------------
region box block 0.0 24.57512 0.0 68.915334 -1.0 58.2455005 units box
create_box 1 box
lattice fcc 3.53848314880901 orient x 1 -1 0 orient y 1 1 -2 orient z 1 1 1
region Co block 0.0 24.57512 0.0 68.915334 -1.0 58.2455005 units box
create_atoms 1 region Co units box
# ---------- Define Interatomic Potential ---------------------
pair_style tersoff
pair_coeff * * CCoW.tersoff Co
neighbor 2.0 bin
neigh_modify delay 10 check yes
# ---------- Create a vacancy by deleting one atom in the center ----------
group DEL_ATOM id 4343
delete_atoms group DEL_ATOM compress yes
# ---------- Assign mass to cobalt atoms ----------
mass 1 58.933
# Minimization
fix 1 all box/relax iso 0.0 vmax 0.001
min_style cg
minimize 1.0e-12 1.0e-12 50000 50000
unfix 1
# Initialize velocities
velocity all create 1000 2547321 mom yes rot yes
# Equilibration
fix 2 all npt temp 1000.0 1000 0.01 iso 0.0 0.0 1.0
thermo 1000
run 200000
unfix 2
reset_timestep 0
# Main run with MSD calculation
fix 3 all nve
compute msd all msd com yes average yes
variable twopoint equal c_msd[4]/6/(step*dt+1.0e-6)
fix 9 all vector 100 c_msd[4]
variable fitslope equal slope(f_9)/6/(10*dt)
fix 4 all ave/time 10 10 100 c_msd[1] c_msd[2] c_msd[3] c_msd[4] file average-msd_Co_1_Vacancy_1000K.txt
# Output settings
thermo 10000
thermo_style custom step temp pe etotal pxx pyy pzz lx ly lz c_msd[1] c_msd[2] c_msd[3] c_msd[4] v_twopoint v_fitslope
log Co_1_Vacancy_1000K.txt
# Dump atom positions for visualization
dump 1 all custom 100000 Co_1_Vacancy_1000K.lammpstrj id type x y z
# Main run
run 3000000
unfix 3
Regards
Fetih
Dear kefyalewfetih
I recently came across your post on the forum about the WC-Co potential function, and I am genuinely impressed by your work. I am currently researching WC-Co alloy systems using LAMMPS, and I have been struggling to find an appropriate potential function. Your contribution is exactly what I’ve been looking for.
You’ve successfully developed a potential function for the WC-Co system, which could be immensely valuable for my research. If it is not too much trouble, would you be open to sharing the potential file or perhaps guiding me on how to write this potential myself? I would be very grateful for any advice or pointers on proceeding with this.
Of course, I fully understand if you are unable to share the file directly, as it is a part of your research. Any suggestions, references, or even just insights into your process would be greatly appreciated and would help me significantly in my work.
Thank you very much for taking the time to read my message. Your Assistance would mean a great deal to me, and I am also happy to share any insights I gain during my learning process.researchingwish you all the best.
Best regards
Yinchun XIAO
Dear 赵辰昊,
Thank you for reaching out to me, and I apologize for the delayed response as I was on leave.
To provide some context, I began my studies with LAMMPS about eight months ago, guided by the invaluable support and suggestions from their representatives. I owe them immense gratitude for their help. During my work, I explored bond-order potential parameters and translated them into a Tersoff potential file.
I reviewed the Tersoff potential file you shared, and I noticed that it is identical to the one I have been using. I attempted to conduct a temperature-dependent study using this potential, but I encountered some challenges during equilibration. Specifically, the system failed to equilibrate properly, and the stress components in all six directions did not converge to zero. Due to these issues, I am currently focusing on simulations at 0 K.
That said, I am actively working to address the equilibration challenges and plan to revisit the temperature-dependent study soon. Once I make progress, I will be more than happy to share my findings and insights with you.
Thank you again for your interest, and I look forward to collaborating further. Please don’t hesitate to reach out if you have any suggestions or questions in the meantime.
Best regards,