I had computer some Potential of Mean Force (PMF) of a polymer collapse with radius of gyration being the collective variable in GROMACS. I am unable to reproduce the same with LAMMPS with the exact same force field settings.
I have tried my best to make sure all the settings are identical, but the PMF from GROMACS and LAMMPS are significantly different and I do not see a reason why they should be different unless there is a difference in the input parameters.
I am attatching my input files for both the simulations. It would be helpful if anyone of you can help me resolve the issue.
The most likely reason are incorrect unit conversions. Please also note that the WHAM code from the Grossfield group has fixed units that must be set at compile time.
The units are correctly taken care of both in PLUMED and WHAM. I compile to different binaries of WHAM one with kcal/mol (for LAMMPS) and one with kJ/mol (for GROMACS). Also, I have noticed that unit conversion issues leads the the PMF being a lot wave-y as wham is not able to stitch together the histograms. However, in this case, the PMFs look completely different (and smooth)
I also specify units in PLUMED when I run LAMMPS simulation
UNITS LENGTH=A time=fs ENERGY=kcal/mol
WHOLEMOLECULES STRIDE=1 ENTITY0=1-32
rg: GYRATION TYPE=RADIUS ATOMS=1-32
restraint-rg: RESTRAINT ARG=rg KAPPA=47.85 AT=$AT
# monitor the two variables and the bias potential from the two restraints
PRINT STRIDE=100 ARG=rg,restraint-rg.bias FILE=COLVAR
Before that is something worth looking into, you need to make certain that forces and energies in both LAMMPS and gromacs are comparable. All of that is not something that you are likely to find anybody to do for you.
Besides, pointing toward units is the only LAMMPS-related advice that can be given without digging deep into your work and re-producing it (which takes considerable time). Provided you can prove without doubt that bother MD engines produce consistent forces, then the difference becomes a question for the Plumed forum at best. The LAMMPS developers do not maintain that code.
A quick note – I understand the desire for a quick solution, but the help you are asking for requires expertise across three separate software packages (LAMMPS, COLVARS, and GROMACS), and not just basic MD knowledge but a specialised area of research (enhanced sampling) and most likely biomolecular simulations (since GROMACS is in the picture).
It’s pretty hard for anyone with that many fingers in that many pies to know how to respond to a first post that essentially says “here’s all my inputs, help!” Even if a reader were infinitely empathetic and kind they would not know where to begin.
As such, please provide a summary of:
what system this is (a “polymer” could be anything from a plastic melt for 3D printing to a coarse-grained model of DNA and histones)
what PMFs you are seeing in LAMMPS and GROMACS? How are they different, and by how much?
what your sampling protocol is (equilibration time, sampling windows of the RC, umbrella spring strength or other sampling parameters) as well as the usual simulation parameters (timesteps and force fields, and with GROMACS in the picture I’d be especially worried about long range electrostatics and neighbourlist settings)
whether one of the PMFs appears “more correct” than another?
I am quite sure that if I did look through your archived files I might be able to determine all of this information. But there are several reasons you should take the time to write out this info:
What if (very unlikely, but) you happened to attach the wrong files, or worse still, files that happened to contain private information?
You need a description like this for your eventual publication any way, or at least to describe your situation to your supervisor.
In the process of writing everything out you may well discover that you made some mistake. Problem located!
Even if you don’t, the process of writing everything out will tell us how you are thinking about the situation (and yourself as well!). This makes things clearer for everyone, making it more likely that we understand where you are coming from, and therefore that what we write can be geared towards your particular situation.
Please do be specific and think about different possible meanings of your sentences – for example, when you write
there are at least three different possible meanings: histograms don’t match up within the GROMACS results, within the LAMMPS results, or between both GROMACS and LAMMPS results.
And as some encouragement – this is a normal process of research. If this were easy and could easily be summarised into a mechanical formula, we wouldn’t need PhDs to do it. Indeed my whole academic career now has come from first finding some code where I couldn’t understand what others had written, and then I couldn’t understand what I had written, to now trying to understand why what I’ve written does what it does. That’s academic research!
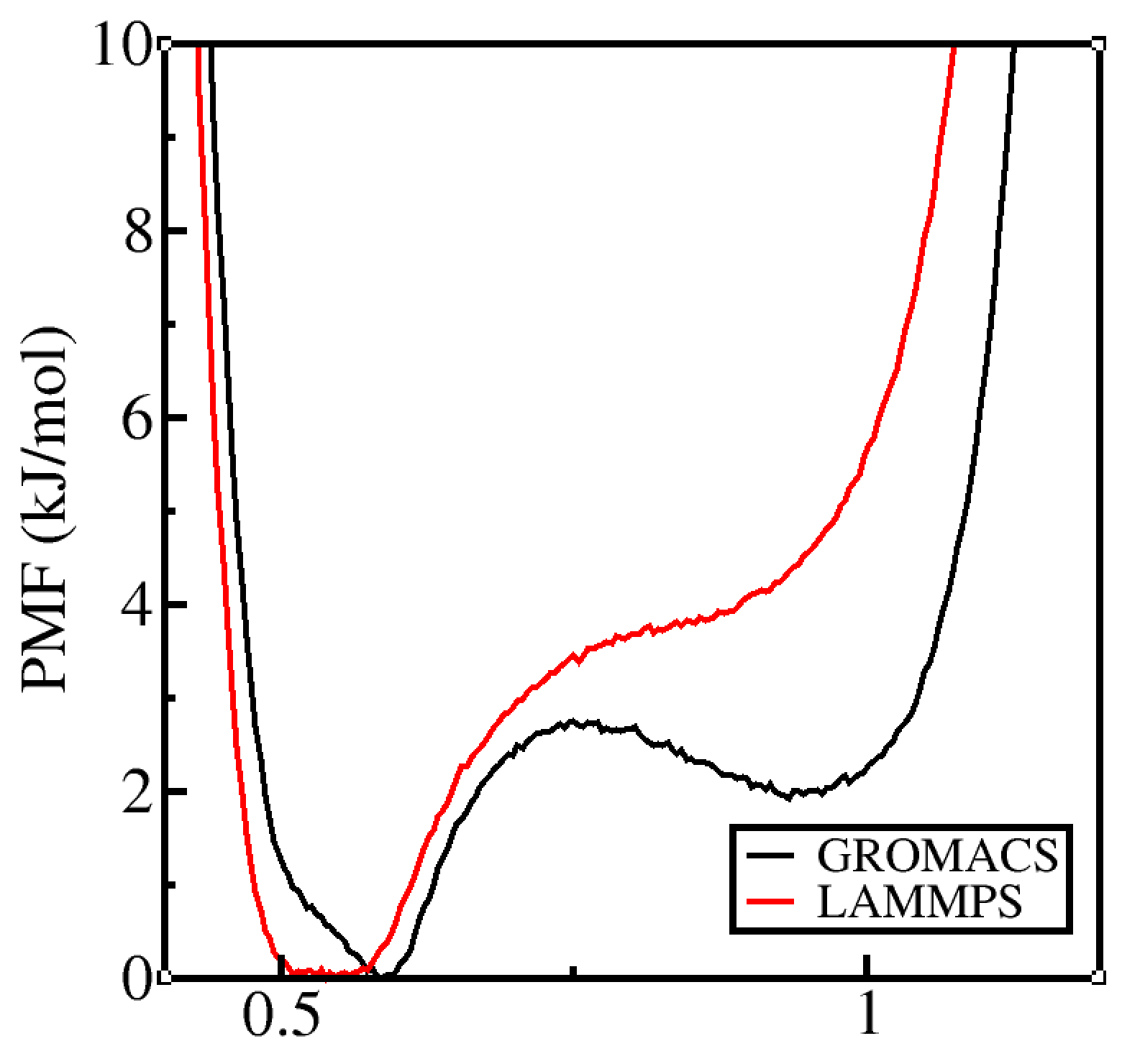
Thank you so much for the message. I apologize for the lack of details. To be precise my system is a model hydrophobic polymer (a bead spring model) with 32 monomers in vacuum. I use Langevin Dynanics to simulate in implicit solvent. The qualitative features of the PMFs do not match between LAMMPS and GROMACS as you can see below:
The polymer is neutral and hence there is no electrostatics involved. Sampling time per window does not change the PMF features. Increasing the sampling time just makes the curve smoother.
As Axel suggested, I looked at the forces that each MD engine gives me, and the forces are of course not exacly the same but they are not two or three order or magnitudes different. Atmost, they are one magnitude higher or lower than the other.
For a single point with the same geometry and after unit conversion, the forces should be close to identical. Your order of magnitude argument is no proof of correct comparison.
One order of magnitude is the difference between a 1.5L bottle of water and a 15kg dumbbell for weightlifting
Relative to such a large difference in forces, the LAMMPS and GROMACS PMFs are similar enough (both essentially quartic with one condensed minimum and another less-condensed minimum).
The most efficient thing to do now is to debug the particle interactions as @akohlmey has said, especially unit conversions and settings. I would recommend the following to make your life simple:
Use just one polymer;
Use NVE integration for both (no point checking thermostatting if the basic interaction potential is different between GROMACS and LAMMPS inputs!);
Use the same timesteps, and force both LAMMPS and GROMACS to update the neighbourlist at set intervals instead of checking (if you know how);
Especially check the 1-2, 1-3 and 1-4 neighbour exclusion settings – I remember these being special_bonds in the LAMMPS input and fudgeLJ in the GROMACS itp files. I recall the defaults being different between the two packages.
The good news is that without electrostatics you should be able to get through this very quickly.
I have found that the issue is certainly not PLUMED as simulations without any bias potential also do not give me consistent results. I like the idea of using NVE simulations and the forces in them are similar:
There are some forces for LAMMPS (first frame)
The order of magnitude difference was a mistake of mine as I did not consder the angstrom to nm conversion.
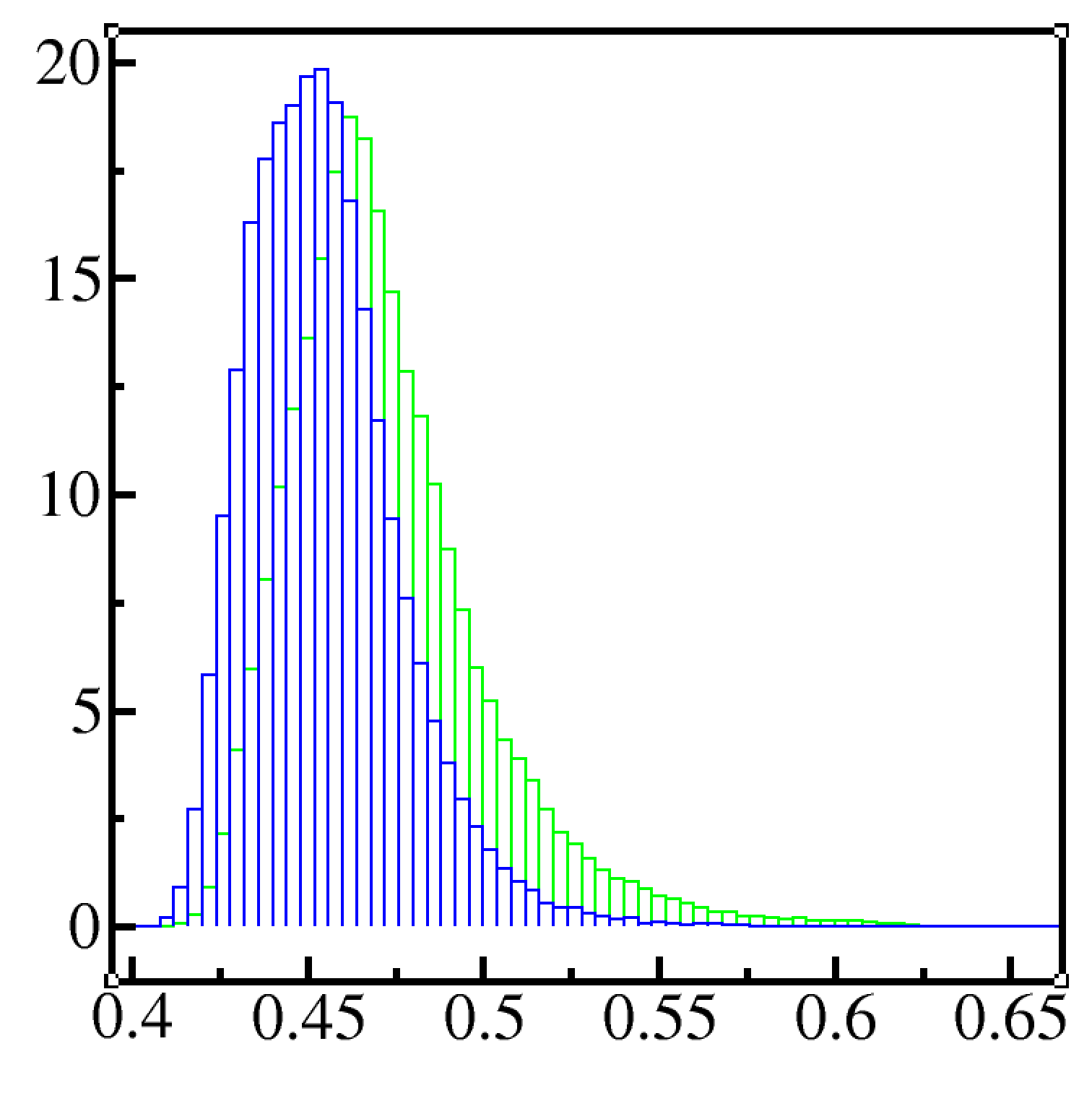
The distribution of Rg (sorry for the labels being absent, but blue is lammps and green is gromacs) tells me LAMMPS gives me more compact structure. My system is just one polymer (32 monomers) in vacuum
In my understanding, a more compact structure can occur if sigma is smaller, bonds are shorter or the angle is smaller. However, those parameters all the same in both my simulations and it would be strange that LAMMPS or GROMACS did not read the values correctly.
I would be careful with such statements. Since both codes have been around a very long time, the chances are very much in favor that you have overlooked something. Since you get different results, you should now check each of the interaction types separately to check if they are all different or only one or two of them. And if that is not sufficient, simplify the geometry to have only a few atoms so that you can compute and sum the force manually.
There you go – good testing (please note this technically does not exclude problems with PLUMED or your PLUMED settings – but one problem at a time, always).
I did ask in my post about:
Please double-check your GROMACS 1-4 settings. This means checking nrexcl under the [ moleculetype ] section in your molecule topology file NAME.top, which might look like:
[ moleculetype ]
; name nrexcl
Urea 3
and the gen-pairs and fudgeLJ settings in the [ defaults ] section in your forcefield.itp file, which might look like:
I have spent fifteen minutes looking through various documentations and still do not understand exactly what GROMACS does (in particular gen-pairs yes seems like it overrides nrexcl 3 to enable 1-4 interactions …), so you will have to test it yourself.
In your position I would just test forces for a single 3-mer and then a single 4-mer as @akohlmey suggested. Takes time, but that’s what you get when you Generate Runs Off Multiple Arcane Collected Scripts …
In GROMACS nrexcl sets the exclusions for non bonded interaction. I have nrexcl = 2 which means that neighbours 2 bonds away are excluded i.e., 1-2 and 1-3 are excluded. gen-pair = yes and fudgeLJ = 0.5 (which is what I use) means that 1-4 interactions are considered and are scaled by 0.5. If I read the documentation for special_bonds, this translates to: special_bonds lj 0.0 0.0 0.5 (which is what I use).
Before even going to a 3-mer, I just checked a 2-mer so its just a dumbell. The forces are all comparabale, and in the first frame are equal.
The following are forces that GROMACS considers (converted to LAMMPS units)
Is your gromacs executable compiled with double precision support? Default is single precision. LAMMPS will use double precision.
At any rate, this discussion has now veered far too far into a territory that has nothing to do with LAMMPS and is about how to validate your work. If you want to discuss on MatSci.org, you can try the Science Talk category, or just consult with your adviser or tutor.
As far as the LAMMPS categories are concerned, I will close this topic now.
You are most welcome to start a new topic, if you have actual verifiable proof that LAMMPS is not doing what its documentation says it will do.