Hi guys, I hope you are fine ^^
I want to simulate the attached model of a Cu NP in Ar using GPU, but I got erratic results. All looks well when I simulate it in CPU with the command:
mpirun --use-hwthread-cpus lmp -in in
and, in my understanding, it should be the same input card for the GPU but executed with the command
lmp -sf gpu -pk gpu 1 -in in
However, the results look very different between both. What do you think?
Thanks a lot!
https://drive.google.com/file/d/1ozgBJKeGdV7sORw_MYhkGrGVl9C0B534/view?usp=sharing
Please provide the following additional information:
- the exact LAMMPS version you are using (or even better provide the output of
lmp -h | sed '/List of individual/Q')
- what kind of GPU you have and how you compiled for it
- if you compiled for CUDA (not HIP or OpenCL), which version of the CUDA toolkit you have used
Thanks, Akohlmey!
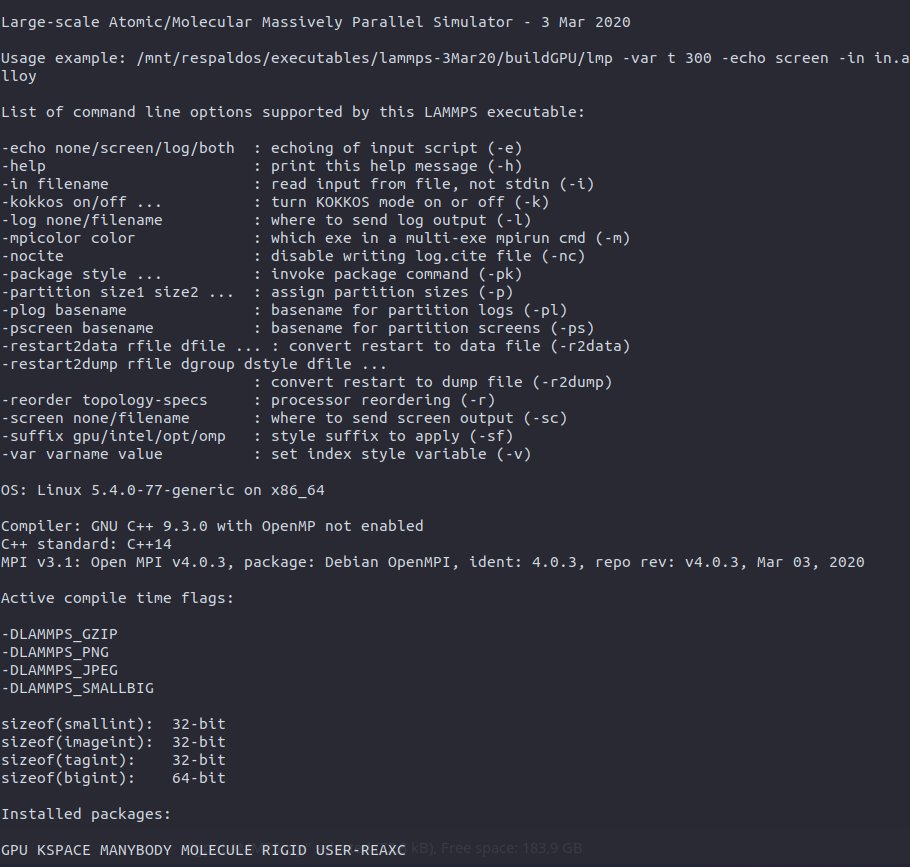
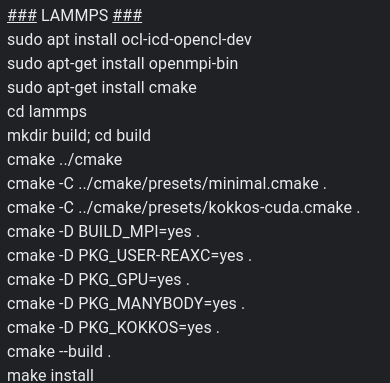
The command returned the attached info. I don’t know if I installed Lammps with CUDA, so I also share the protocol that I used. Anyway, here I read that my gtx 150 maxq is Cuda capable.
Thanks a lot one more time.
@GabrielOlguinOrellan thanks for the info.
You compiled LAMMPS with OpenCL, not CUDA. This should be reported on your screen.
But that is not very important in this case since your LAMMPS version is a bit older.
The main problem here is that using hybrid atom styles with the GPU package don’t always support using the neighbor list build on the GPU. This is a known problem that is still not yet fully resolved: [BUG] Using pair style hybrid with GPU package and neighbor lists on GPU · Issue #2621 · lammps/lammps · GitHub
So please try the following following command line to run LAMMPS. That should give you (mostly) the same output (it is different because the GPU package uses by default a mix of single and double precision while the CPU always uses double precision) and a correct trajectory:
lmp -sf gpu -pk gpu 1 neigh no -in in
Thanks a lot, Axel! your suggestion solved my problem as many other times from the email chain.
Regarding my installation, you would suggest upgrading my Lammps? install it with CUDA? I’m also interested in simulating a system including graphene, with parameters from AIREBO, and also with Reax/c.
As I understand, I can’t use the GPU package for AIREBO or Reax/c, so I should install Kokkos, but I’m a bit afraid of breaking my current drivers installation in the process. Could you give us some suggestions to accomplish it?
Thanks a lot one more time.
Greetings from the Center for Bioinformatics, Simulation and Modeling, Chile.
Would you mind posting your followup question as a new question in the “LAMMPS Installation” category (where it would be a better fit)?
1 Like