Dear all,
I am simulating the stretch of interlocked ring molecules, and I used the bead-spring FENE model with LJ units. But the rings easily cross each other when I stretch two ends of the chain. Are there any suggestions about this? I don’t think it is normal, so how to prevent the ring crossing in the bead-spring model simulation?
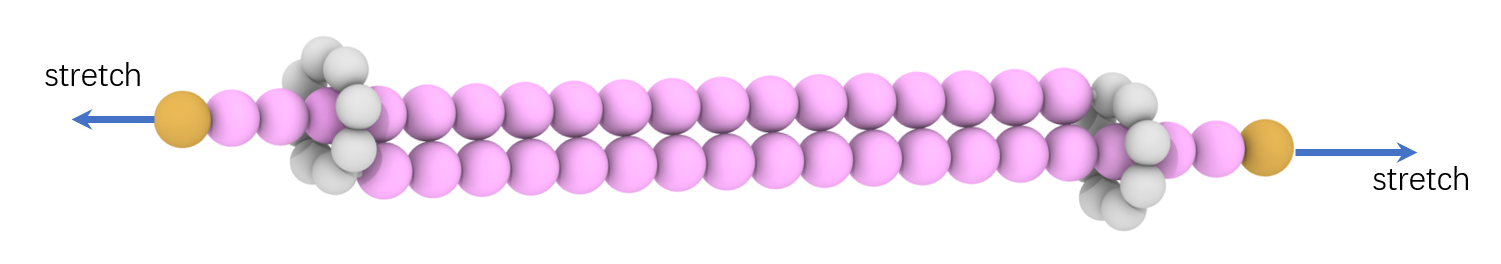
I posted the snapshot of the interlocked ring molecule, the stretching video and the main lammps script below:
##################################################################

########################## Initialization ######################
units lj
boundary p p p
atom_style fullbond_style fene
angle_style cosine
pair_style lj/cut 2.5
special_bonds feneneighbor 1 bin
neigh_modify every 1 delay 0 check yes#################################################################
read_data DaisyChain.data
#################################################################
mass * 1bond_coeff * 30.0 1.5 1.0 1.0
angle_coeff 1 1000.0 # ring
angle_coeff 2 1.0 # axle#################################################################
pair_coeff * * 1 1 0 2.5
pair_coeff 4 * 1 1 1 2.5
pair_coeff 3 5 5 1 0 2.5
pair_coeff 1 6 5 1 0 2.5
pair_coeff 1 3 1 1 1 2.5##################################################################################
####################################################################
timestep 0.005
variable TH equal 1.5
variable T equal 0.4
variable Random equal “300*round(random(10, 5000, 424))”##################### Equilibration ######################
reset_timestep 0
velocity all create {T} {Random}
minimize 1.0e-6 1.0e-6 10000 10000thermo 100
thermo_style custom step etotal press vol temp density pxx pyy pzzdump 11 all custom 2000 relax.lammpstrj id mol type x y z
fix fix1 all nvt temp {T} {T} 1
run 20000
unfix fix1
undump 11######################## Deformation #################################
run 0
reset_timestep 0################# Define Variables ###################
variable yl equal “ylo+1”
variable yh equal “yhi-1”
variable yh2 equal “yhi*3”
variable y_step equal “yhi/2”variable p3 equal “ly”
variable L0 equal ${p3}
variable strain equal “(ly - v_L0)/v_L0”
variable p1 equal v_strain
variable p2 equal “-pyy/10”#################### define groups ###################
group wall id 54
group pull id 27
group boundary union wall pull
group mobile subtract all wall #pull#################### computes ###################
compute peratom all stress/atom NULL pair#################### initial velocities ###################
velocity all create 1 887723#################### fixes ###################
fix fix1 all nvt temp {T} {T} 1
fix f3 wall setforce 0.0 0.0 0.0
fix f4 pull setforce 0.0 0.1 0.0#################### Dump ###################
dump 11 all custom 10 tensile.lammpstrj id mol type x y z
dump 100 all custom 1000 stress_per_atom.txt id type x y z c_peratom[1] c_peratom[2] c_peratom[3] c_peratom[4] c_peratom[5] c_peratom[6]
fix def1 all print 100 “{p1} {p2}” file Stress_strain.def1.txt screen nothermo_style custom step temp pyy lx ly lz v_strain
thermo 100
thermo_style custom step temp vol press ke pe lx ly lz
timestep 0.01
run 30000write_data final.data
print “All Done”