Hello all,
I am attempting to simulate equilibrium (i.e., quiescent) monodisperse hard-sphere fluids using the Fast Lubrication Dynamics (FLD) treatment of hydrodynamics, where I am overlaying purely-repulsive 48-24 Weeks-Chandler-Andersen potentials (to approximate the hard-sphere interactions) on top of lubricate/poly and brownian/poly pair-styles. (Here, I am using ‘poly’ because I want to generalize in the end to ternary systems; for now the three species all have sigma=1)
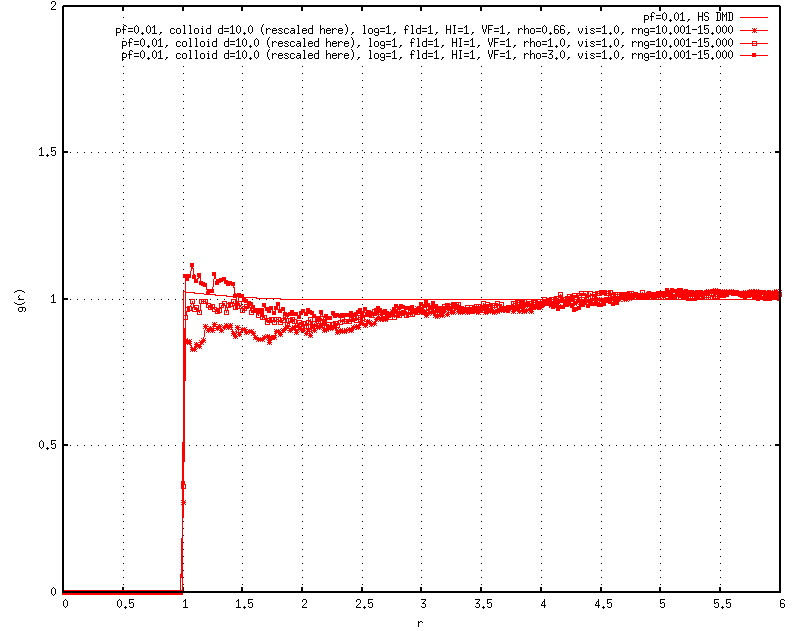
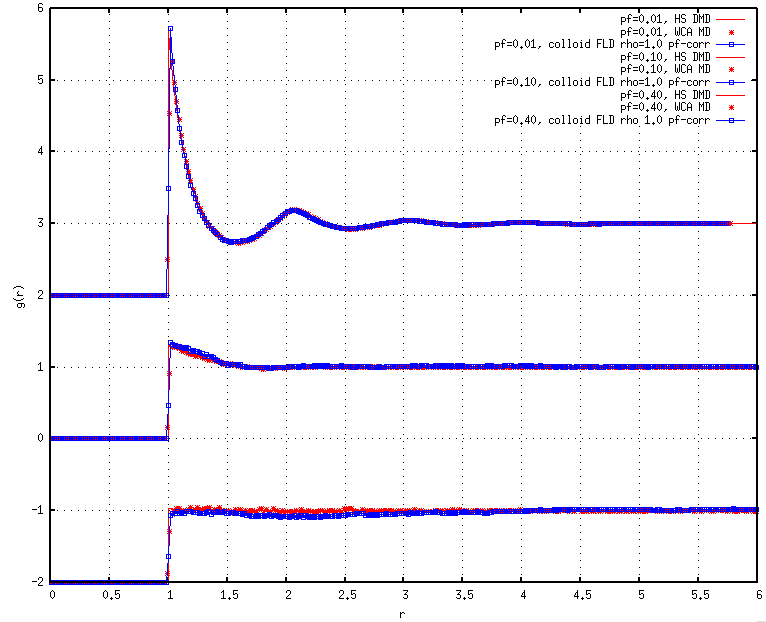
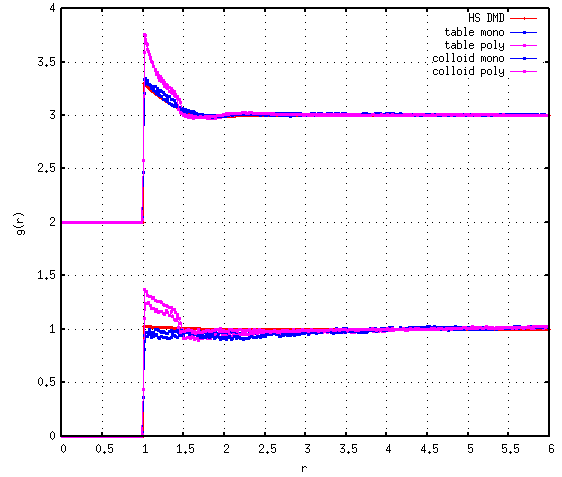
As I began to test these HS systems at different moderate packing fractions, I noticed that the radial distribution functions (RDFs) did not match previous results for WCA fluids obtained using NVT MD, instead exhibiting stronger contact values and signatures that seemed to be shifted upwards for the first and/or second coordination shells, perhaps indicating net attractions.
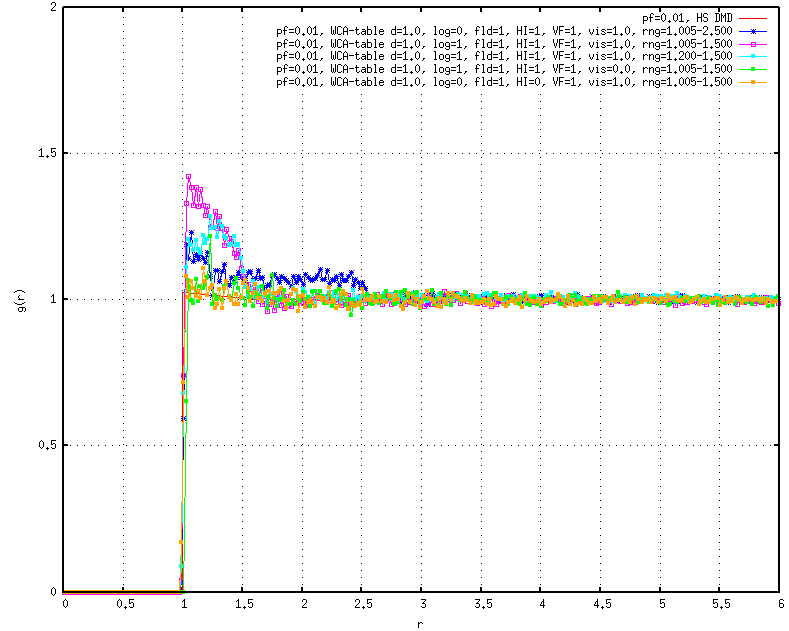
In turn, I decided to simulate a low-density system (packing fraction = 0.01), which should exhibit a step function RDF in equilibrium. Instead, I find that there are net attractions (indicated by peaks in the RDFs) between the inner and outer cutoffs of the lubrication interactions, which I have confirmed by trying different combinations of cutoffs (1.001-1.500, 1.200-1.500, 1.300-1.500, 1.001-2.500, etc.). These are not small effects, either–e.g., using cutoff 1.001-1.500, I get a contact value at r=sigma of 1.5, a peak that then decays completely by just outside of 1.500.
These effects emerge whether flaglog is turned on or off (i.e., including or excluding the Ball and Melrose log-terms), though the “attractions” are stronger when flaglog in turned on. In all cases I have tried, I have kept flagFLD, flagHI and flagVF on.
At this point, I’m a bit at a loss. Even given I am misusing the FLD commands (or even if there is a bug), I would tend to expect depletion deviations rather than attractive. Visualizing the systems does not point to anything fishy, and I should note that the RDFs do not point to any particle overlaps.
Has anyone had an issue like this previously? Any guidance/ideas would be greatly appreciated.
Details: I am using lammps/2014_01_10 run in parallel on 16 processors. Input files are listed below:
units lj
dimension 3
boundary p p p
atom_style sphere
newton off
communicate single vel yes
pair_style hybrid/overlay lubricate/poly 1.0 1 1 1.005 1.500 1 1 brownian/poly 1.0 1 1 1.005 1.500 1.0 42115 1 1 table spline 1500
read_data file.data
neighbor 0.2 bin
pair_coeff * * lubricate/poly
pair_coeff * * brownian/poly
pair_coeff * * table wca.table WCA 1.20000
thermo_style custom step xy temp ke pe etotal press
thermo 1000
timestep 0.0002
fix 1 all nve/sphere
dump xtcdump all xtc 1000 out.xtc
dump_modify xtcdump precision 100000
restart 1000000 restart.*
log log.lammps
run 1000000
unfix 1
Along with the file.data:
[comment line]
1200 atoms
3 atom types
0.0 38.51197 xlo xhi
0.0 38.51197 ylo yhi
0.0 38.51197 zlo zhi
0.0 0.0 0.0 xy xz yz
Atoms
1 1 1.0000 1.9099 15.0300 24.6310 31.6730
2 1 1.0000 1.9099 38.2340 30.3260 6.5280
3 1 1.0000 1.9099 18.9990 7.8190 32.7410
…
1200 3 1.0000 1.9099 31.2680 29.4610 17.0900
WCA potential file not including for now for length’s sake–rest assured I checked to ensure it is correct.