Dear Mr.mattmcdermott,
Thank for your great information,
I was use te code for calculate the electrochemical inferface energy between solid elctrolyte|cathode as bellow:
from pymatgen.ext.matproj import MPRester
from pymatgen.analysis.interface_reactions import GrandPotentialInterfacialReactivity
from pymatgen.analysis.phase_diagram import PhaseDiagram, GrandPotentialPhaseDiagram, PDEntry, PDPlotter
from pymatgen.core import Composition, Element
%matplotlib inline
mpr = MPRester('API')
reactant1 = 'LiCoO2'
reactant2 = 'Li3PS4'
grand = True
if grand:
open_el = 'Li'
relative_mu = -2
comp1 = Composition(reactant1)
comp2 = Composition(reactant2)
elements = [e.symbol for e in comp1.elements + comp2.elements]
if grand:
elements.append(open_el)
elements = list(set(elements))
entries = mpr.get_entries_in_chemsys(elements)
pd = PhaseDiagram(entries)
if grand:
mu = pd.get_transition_chempots(Element(open_el))[0]
chempots = {open_el: relative_mu + mu}
gpd = GrandPotentialPhaseDiagram(entries, chempots)
interface = GrandPotentialInterfacialReactivity(
comp1, comp2, gpd, norm=True, include_no_mixing_energy=True, pd_non_grand=pd, use_hull_energy=False)
else:
interface = GrandPotentialInterfacialReactivity(
comp1, comp2, pd, norm=True, include_no_mixing_energy=False, pd_non_grand=None, use_hull_energy=False)
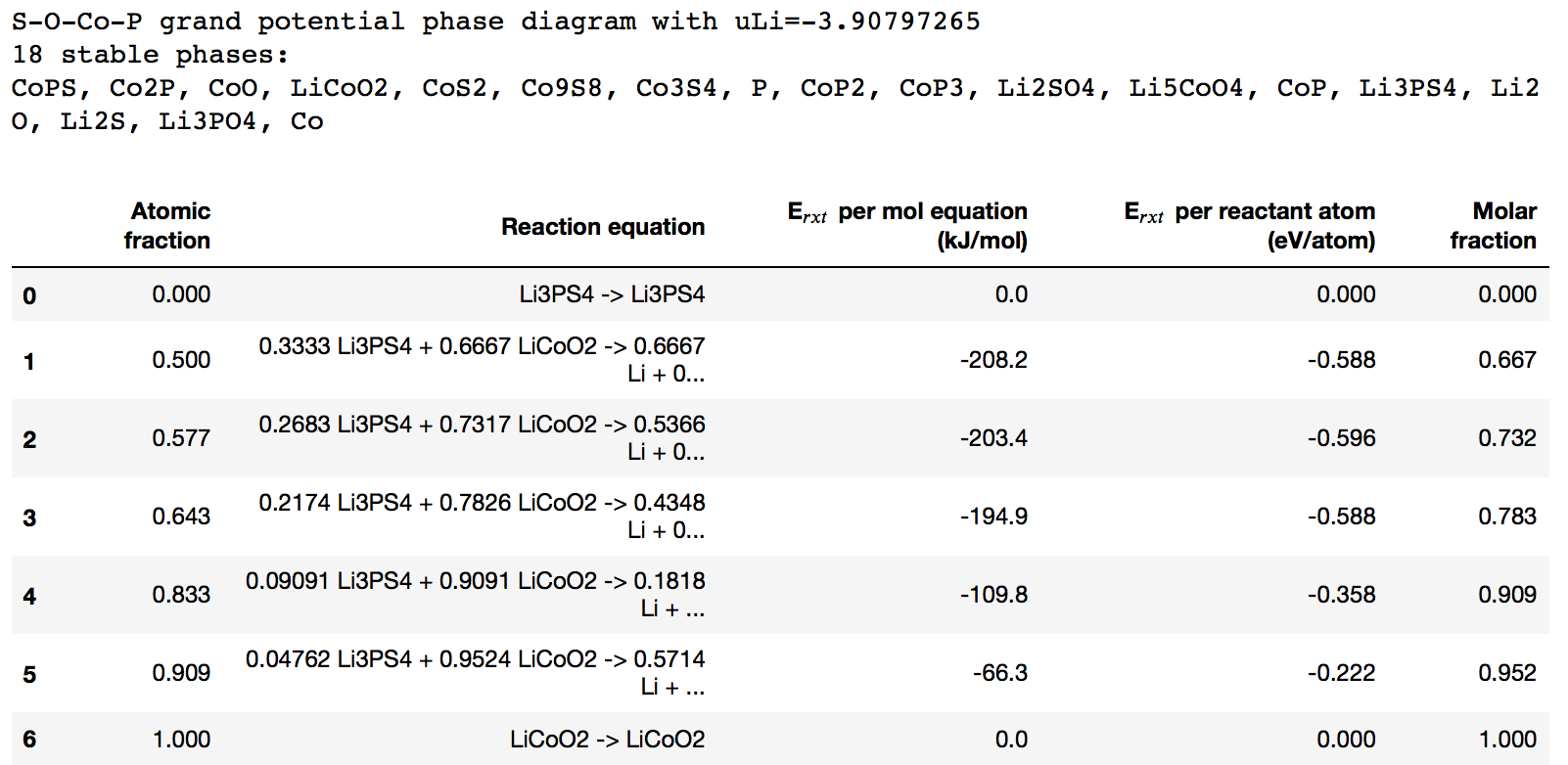
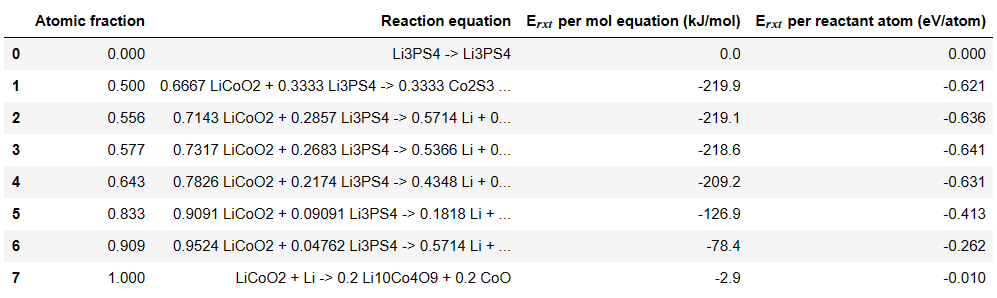
interface.plot()
from collections import OrderedDict
import pandas as pd
critical_rxns = [
OrderedDict([
("Atomic fraction", round(ratio, 3)),
("Reaction equation", rxn),
("E$_{rxt}$ per mol equation (kJ/mol)", round(rxn_energy, 1)),
("E$_{rxt}$ per reactant atom (eV/atom)", round(reactivity, 3)),
])
for _, ratio, reactivity, rxn, rxn_energy in interface.get_kinks()]
interface_reaction_table = pd.DataFrame(critical_rxns)
interface_reaction_table
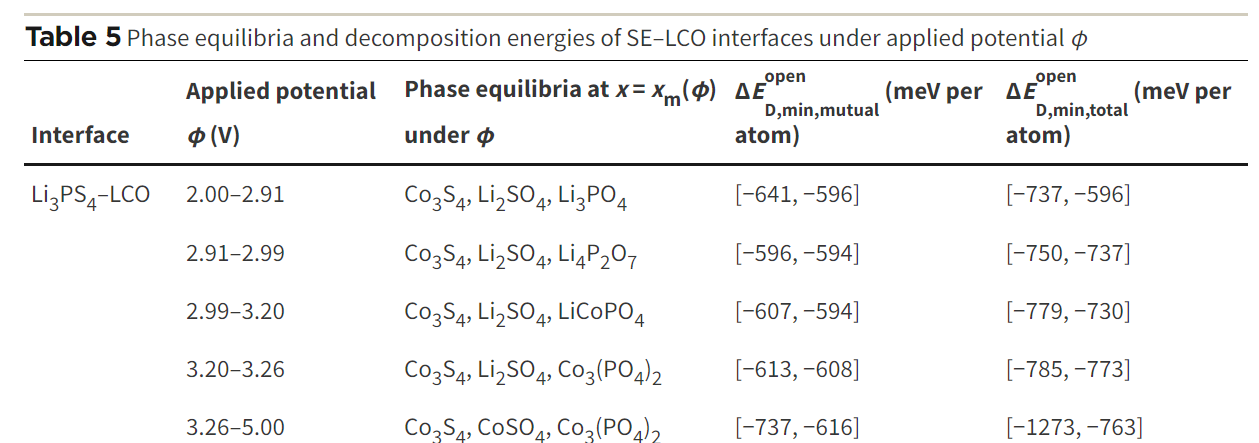
I am follow this paper (First principles study on electrochemical and chemical stability of solid electrolyte–electrode interfaces in all-solid-state Li-ion batteries - Journal of Materials Chemistry A (RSC Publishing))
With mu_Li=-2 , the code print minimum energy reaction is -641 meV/atom. However, when I apply mu_Li = -2.91 the code print minimum energy reaction is -783 meV/atom, it is different value with table 5 on paper. So I can not understand the meaning of mu_Li and the chempots in the code.
How can I apply potential of charge bateries for calculate electrochemical interface reaction?
Thank you so much for helping,
Best reagrds,
Tham