With the help of the widget, I converted the crystal structure model from the CAR file in Material Studio to the LAMMPS data file and imported it into LAMMPS. When I used lammps relaxation, I found that the crystal density became much larger, it should be 1.5~1.6, and the result became 1.9, which is 25% larger. But when this crystal structure is dynamic in Material Studio, the density is normal, how can I make the density normal in the lammps (it is normal before relaxation, and it can’t be so large)?
Is it possible to increase the box size directly in lammps, or increase the value in the z direction?
Hi,
I am unfamiliar with Material studio, which I believe is out of the scope of this forum, but the question is not very clear. Which kind of relaxation are you performing. Which constraints are you using? Are you barostating the system to 0 pressure using any NPT ensemble?. Which material is it and which interatomic potential are you using. When I perform any crystal relaxation I start by minimizing the system using min_style cg in conjunction with fix box_relax. This should tell you also about the stability of your configuration.
When you get a different density with two different simulation software packages, you must have made some mistake(s) setting up the topology or force field parameters or simulation settings in either of the two software packages (or both).
Yes, there is the change_box command, but that won’t resolve the problem(s) I mentioned.
Thank you very much for your reply. I’m using pcff force field on both sides, and the error you raised is setting up the topology or force field parameters or simulation settings in either of the two software packages (or both).;; Can you elaborate on what parameters might affect the density change, I would like to solve the fundamental problem.
The model is the foundation, very important to me, what information you need I am willing to provide you.
At the very least show us your input.
ok,I show you the structure and density in Material Studio; How I converted, and the density after the conversion.
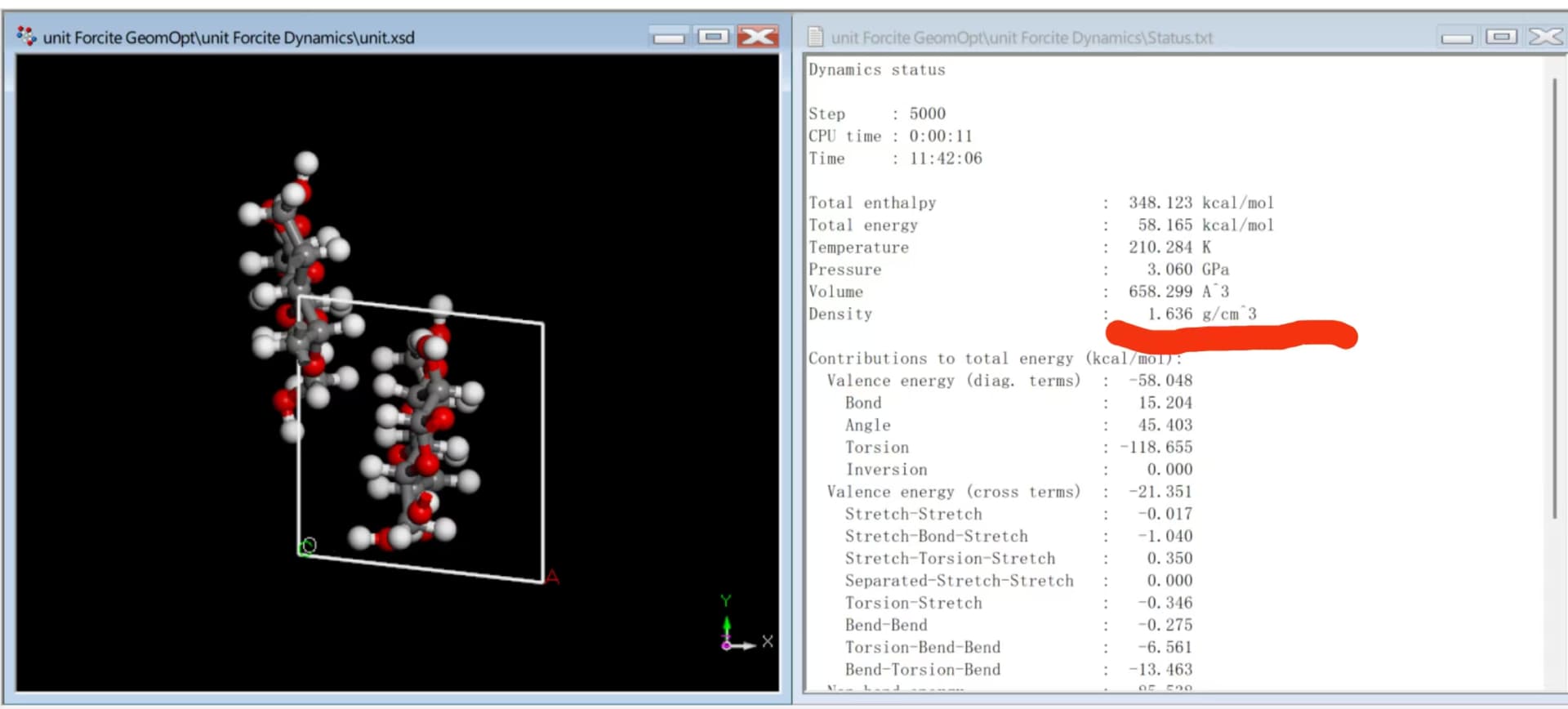
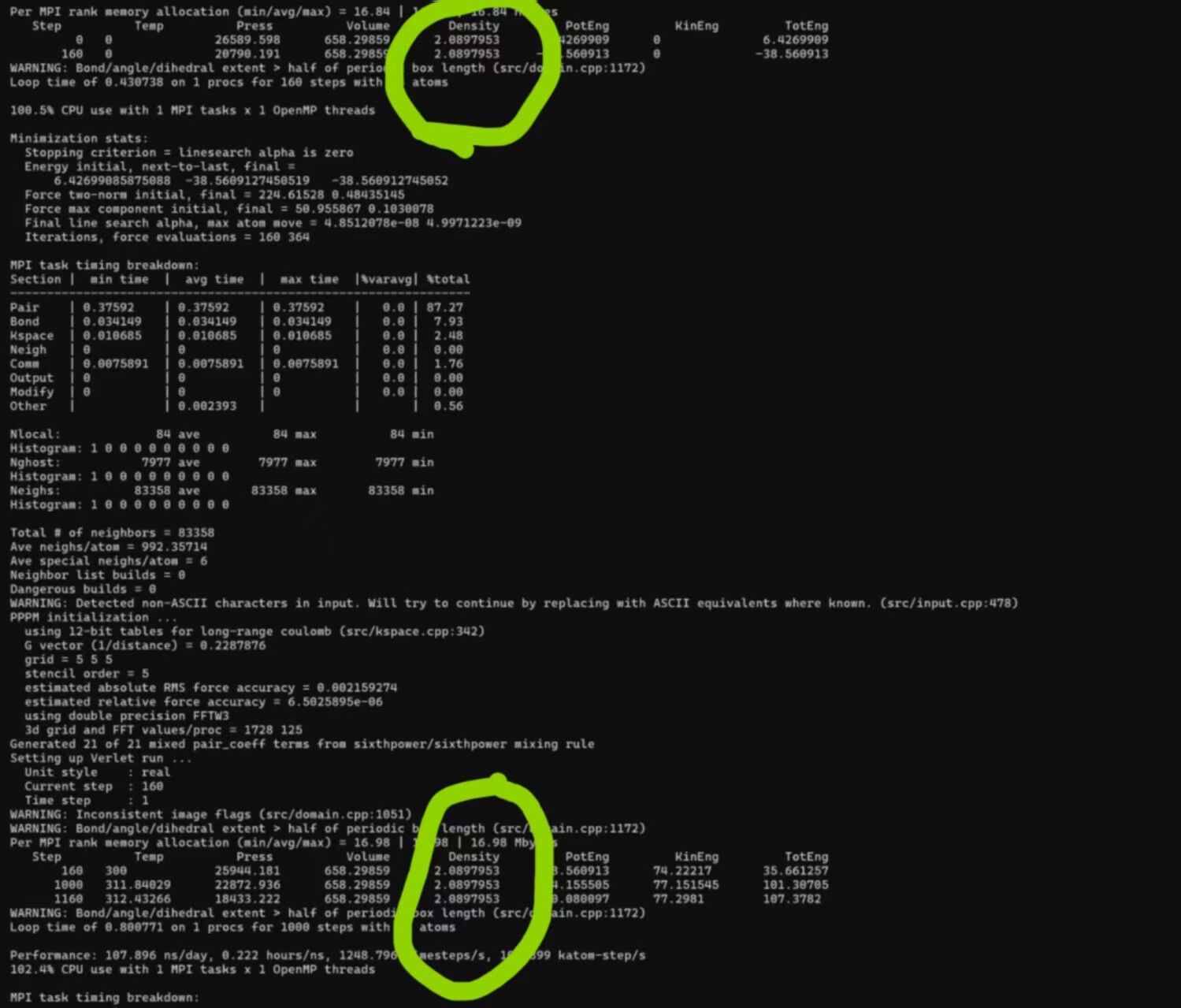
insight2lammps.pl (39.1 KB)
pcff.frc (332.4 KB)
The last two documents are from EMC.
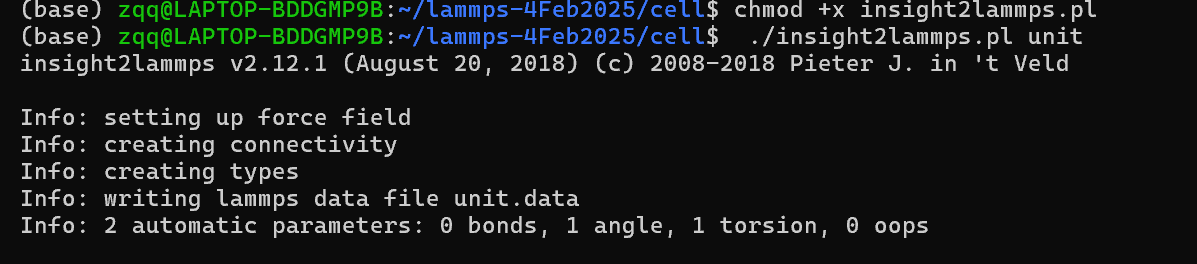
This is the conversion operation process, before the conversion I changed the ‘‘compass’’ in the insight2lammps.pl to ‘‘pcff’’.As shown in figure.

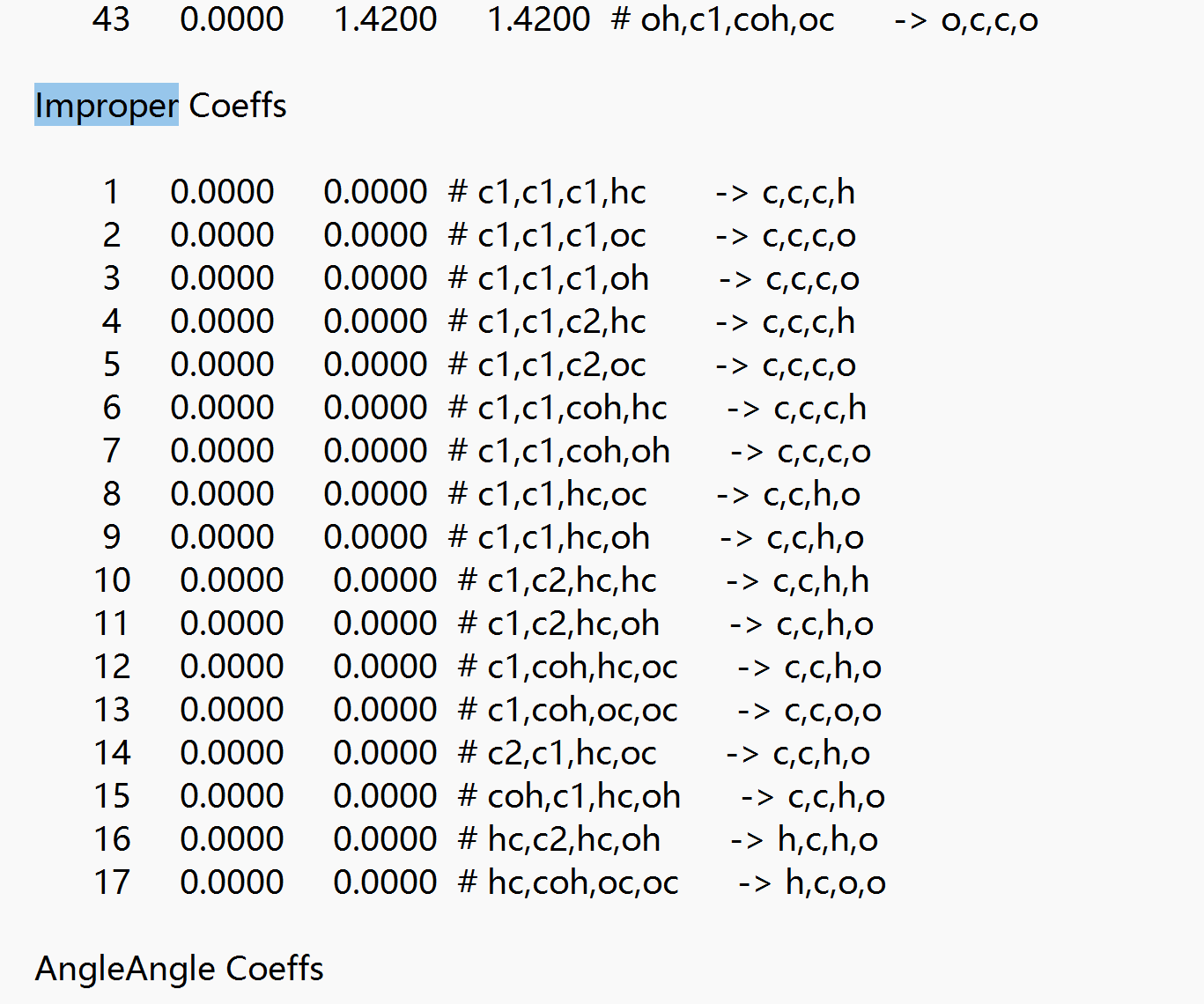
This part of my data file is 0.My structure is a cellulose crystal structure, what is causing this problem。
thank you for your reply.In my data file,my improper Coeffs show zero.Could this be the reason for the inconsistency in density?
It is not possible to work out what your actual process was from the screenshots you have given.
But: take a look at your LAMMPS screenshot. The volume has remained the same all through your LAMMPS calculation. Wherever the issue has occured it is therefore before you even started running LAMMPS – probably an incorrect conversion between the output format of the previous program and the input format of LAMMPS.
It is not possible for us to diagnose any further than that.
1 Like
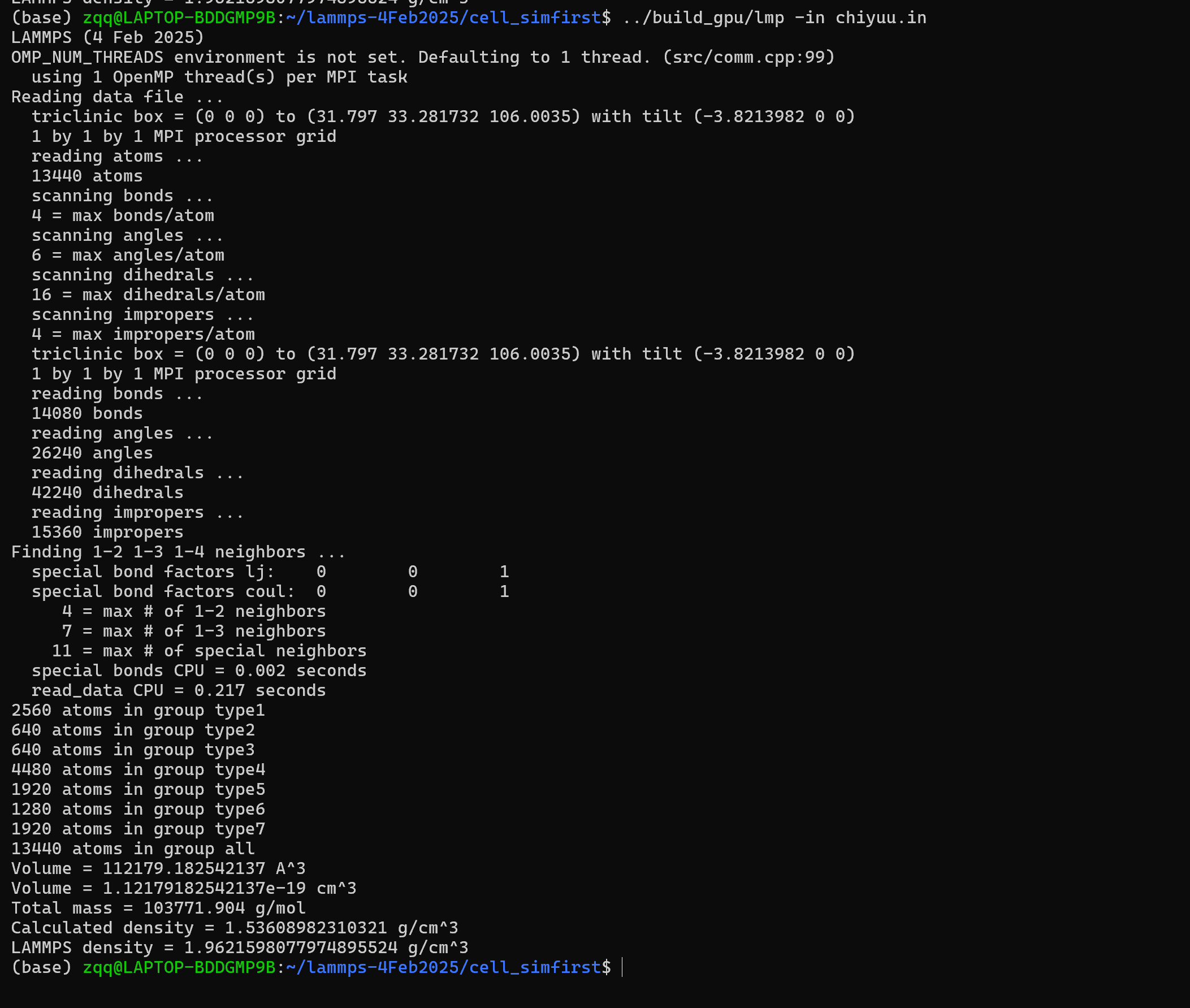
Thank you very much for your reply, the first one is the previous Chinyu.in file. I checked again, and it should be that lammps calculates density differently than material studio, and when I add this, I write chiyuyu.in file. The output is the same as in Material Studio
Chiyu.in (2.0 KB)
chiyuu.in (3.1 KB)
group type1 type 1 # c1
group type2 type 2 # c2
group type3 type 3 # coh
group type4 type 4 # hc
group type5 type 5 # ho
group type6 type 6 # oc
group type7 type 7 # oh
创建包含所有原子的组
group all type 1:7
计算每种原子的数量和质量
variable n1 equal count(type1)
variable n2 equal count(type2)
variable n3 equal count(type3)
variable n4 equal count(type4)
variable n5 equal count(type5)
variable n6 equal count(type6)
variable n7 equal count(type7)
定义阿伏伽德罗常数
variable NA equal 6.02214076e23
计算总质量 (g/mol)
variable total_mass equal 12.01115v_n1 + 12.01115v_n2 + 12.01115v_n3 + 1.00797v_n4 + 1.00797v_n5 + 15.9994v_n6 + 15.9994*v_n7
体积转换
variable vol_A3 equal vol # 体积(ų)
variable vol_cm3 equal v_vol_A3*1.0e-24 # 转换为 cm³
计算正确的密度(考虑阿伏伽德罗常数)
variable calculated_density equal ${total_mass}/(v_NA*v_vol_cm3)
输出信息
print “Volume = {vol_A3} A^3"
print "Volume = {vol_cm3} cm^3”
print “Total mass = {total_mass} g/mol"
print "Calculated density = {calculated_density} g/cm^3”
print “LAMMPS density = $(density) g/cm^3”
Looking forward to hearing from you