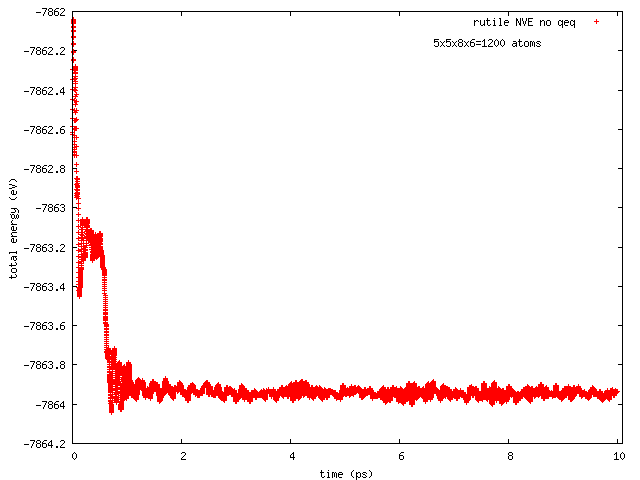
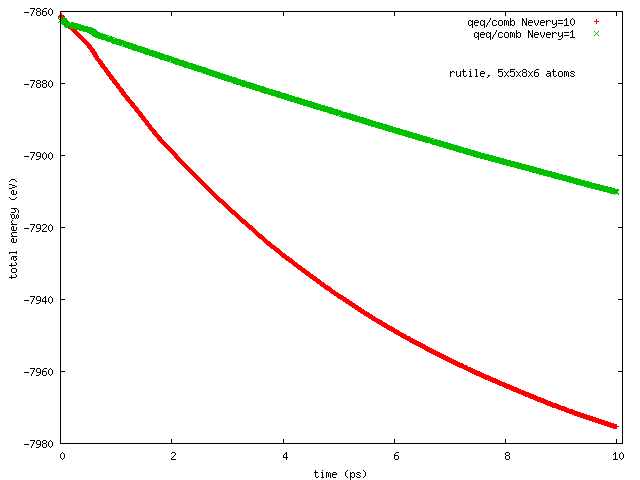
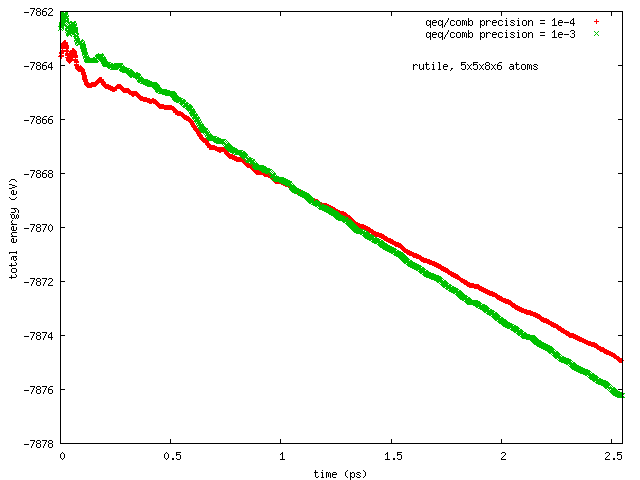
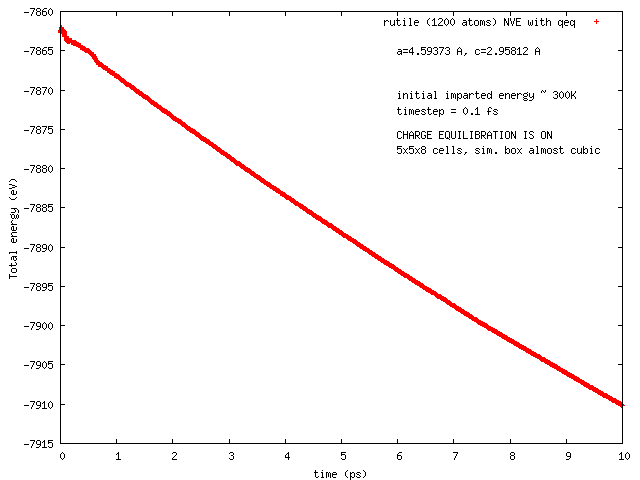
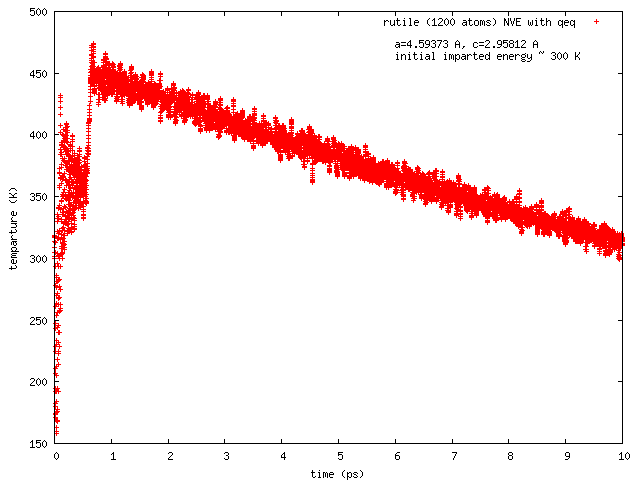
Hi all, this has been reported earlier by Arthur France-Lanord. The issue is total energy does not remain conserved in NVE runs with COMB3 potential while charge equilibration is on (NVE+qeq/comb). The total energy decreases progressively, and so does the temperature.
Pls find attached two plots of total energy and temperature (vs time) from a NVE run with rutile (TiO2). I used the comb3 potential that is provided in lammps distribution (stable version of 1 Feb 2014). Pls also find attached the files required to run this on lammps (in.*, structure, potential: ffield.comb3, taken from lammps distribution).
The rutile structure file has been inspected carefully, and is hopefully correct. Per Tao Liang’s suggestion, the system size has been so chosen that each dimension is equal or greater than twice of the cutoff length for Coulomb interaction set in comb3 (which is 11 A, the 35th element in any entry in ffield.comb3, and read as params[nparams].coulcut in pair_comb3.cpp). The lammps input file is a modified version of one I received from Ray Shan earlier.
Is there anything in the mathematics of charge equilibration (Rappe and Goodard 1991, Rick and Stuart 1994) that bars the total energy from being conserved in NVE in comb3? I was curious to know what is the scenario in case of qeq/reax (which uses Nakano 1997). I need to build lammps with relevant packages to check that. Meanwhile, any comment on all these will be much appreciated.
Best regards.
– Abrar
in.rutile_NVE (814 Bytes)
ffield.comb3 (324 KB)
data.rutile_5x5x8 (90.7 KB)