Dear all, hope you are well and wiil have a nice holiday soon.
I am trying to do a calibration test for my clump, therefore hope to use a rigid wall to compress my clump to obtain the force - displacement relationship.
Following the very useful suggestions from experts:
i decided to use fix wall/gran with wiggle to shear my clump.
however, i have a weired problem that i cannot actually see the contacts between my clump and the wall.
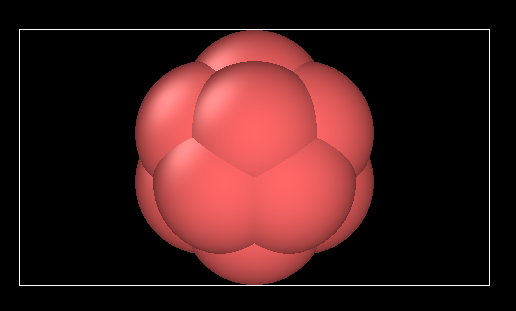
i generated this clump with top boundary 0.01261; bottom boundary -0.00501. as illusrated in the image, they are close to clump but not really contacting the clump
fix 3 give the bottom boundary close to clump, and fix 4 give the top boundary close to the clump. at the same time. i used the wiggle to compress this clump. The amplitude is 0.1 m, which should be large enough. however i cannot identify the contacts between clump and wall.
this is confirmed by
There is far too much going on in your input to make a suggestion by just looking at it.
What stands out is that there is no time integration, so atoms don’t move. If this is added, the system cannot run without either losing atoms or bonds, which suggests that there is something fundamentally wrong with your model. However, I have too little experience with DEM simulations to know any specifics.
If you want somebody to seriously debug this, you need to simplify your input, e.g. just have a few individual particles without bonds just using atom style sphere and only a pair style. Excessive use of variables like in your input also - while convenient - make it difficult to debug because one always has to look up which number a value represents. So for debugging, it would be better to not use them. If you do get the expected behavior (contacts being recorded) with the simplified input, you can then incrementally add “complications” and then you will notice when a command introduces an instability (i.e. lost atoms/bond atoms) or loses the desired feature (reporting contacts). Depending on the outcome of that one can further discuss whether there is a problem with your model or with the functionality of LAMMPS.
Many thanks for your reponse again. i finally get the potential reason why i cannot identify the contacts between the wall and particles.
for the losing atoms or bonds. I will follow you suggestions and try to use may be just one single sphere also without variable to see whether i can observe the contacts between wall and spheres.
hope you have a nice holiday
Just for anyone who may interested in the reason why getting losing atoms or bonds for bpm package;
the significant small timp step is required; that is 1e-8 or 1e-9 depending on the parameters.