Dear user,
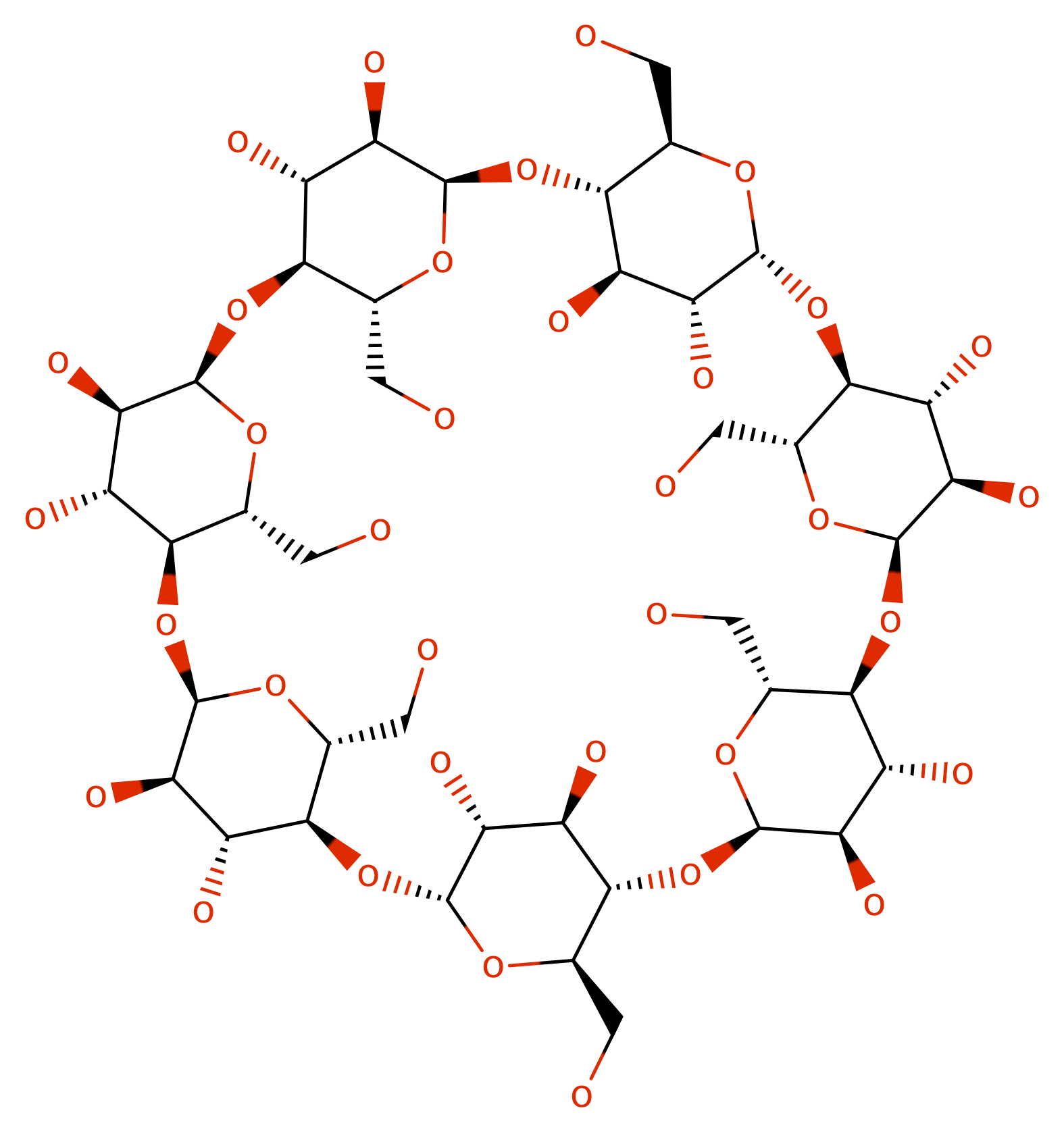
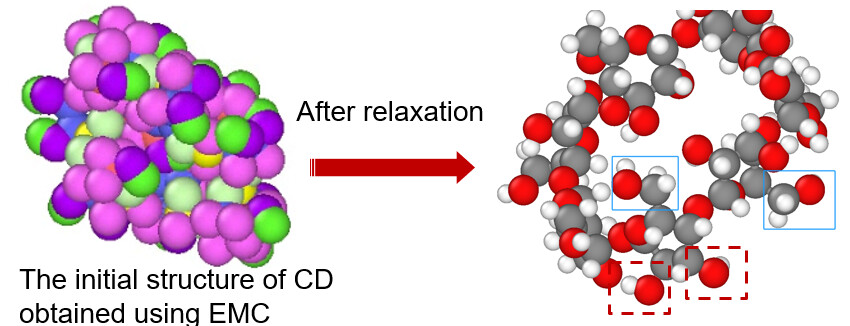
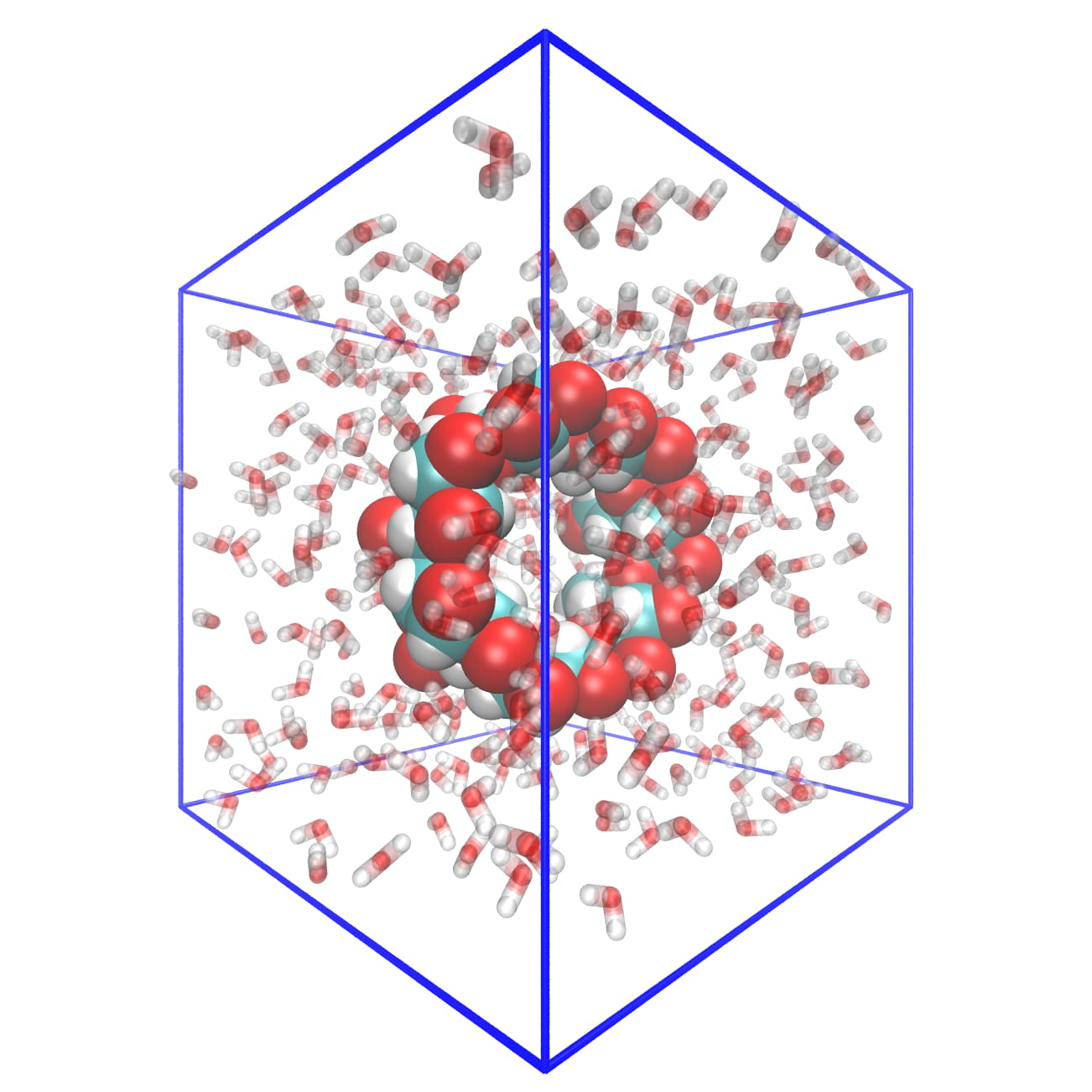
Your chemistry is a carbohydrate super structure, which consists of seven six-membered rings and one overarching 42-membered ring, as shown in the following
EMC uses a scanning depth to recognize these rings, which by default is set to 8. In order to be able to build your system, you would need to increase this scanning depth to at least 42 – I would take 50 to be safe – which can be done by adding
emc_depth 50 to your
ITEM OPTIONS block. The resulting
.esh script would read
#!/usr/bin/env emc_setup.pl
ITEM OPTIONS
replace true
field pcff
density 0.01
number true
emc_depth 50
emc_execute true
ITEM END
# Groups
ITEM GROUPS
mol C([C@@H]1[C@@H]2[C@@H]([C@H]([C@H](O1)O[C@@H]3[C@H](O[C@@H]([C@@H]([C@H]3O)O)O[C@@H]4[C@H](O[C@@H]([C@@H]([C@H]4O)O)O[C@@H]5[C@H](O[C@@H]([C@@H]([C@H]5O)O)O[C@@H]6[C@H](O[C@@H]([C@@H]([C@H]6O)O)O[C@@H]7[C@H](O[C@@H]([C@@H]([C@H]7O)O)O[C@@H]8[C@H](O[C@H](O2)[C@@H]([C@H]8O)O)CO)CO)CO)CO)CO)CO)O)O)O
ITEM END
# Clusters
ITEM CLUSTERS
mol mol,1
ITEM END
# EMC
ITEM EMC phase=1 spot=2
test_chirality = {
mode -> true
};
ITEM END
I added number true to avoid having to provide ntotal. This option means, that the number at the end of mol mol,1 is interpreted as the number of molecules or clusters to build. I also added a verbatim EMC Script paragraph to test chirality of your structure. This paragraph is added right after the build in build.emc and produces the stereo chemical state. It allows you to see, which stereo chemical centers stay the same, and which change after multiple builds.
A word of caution with respect to stereo chemistry: EMC produces correct stereo chemistry for linear molecules. However, it makes mistakes when considering ring structures like sugars when the ring closing atom is concerned. This ring closing atom is defined by your SMILES. I am aware of the issue and have a route to solution, but unfortunately haven’t had the time to fix it, since the fix is rather involved. Please decide yourself, if correct stereo chemistry is needed for the model you are developing. In the above case, I would roughly estimate, that 75% of the stereo chemistry is correct.
The above example can also end up not being build. This happens either because ends are not found or the molecule is growing too much into itself. Three settings influencing the build procedure are niterations, radius, and nrelax, of the which the defaults are 1000, 5, and 100 respectively. The niterations option sets the number of trials to find a solution before aborting. One could try to increase the number of iterations. The radius option controls the radius within atoms are relaxed. The center of the sphere is the added atom. I would not go higher that 10 Angstrom. nrelax controls the number of relaxation cycles EMC uses. Upping this number will result in more relaxed structures. Increasing either radius or nrelax or both will slow down the building.